

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Lamins are the major components of the nuclear lamina, a network located between inner nuclear membrane and chromatin, which plays a fundamental role in the organization of the nuclear architecture in all human cells. Lamins A and C, which are alternatively spliced products of the A-type lamin gene (LMNA), are expressed in differentiated cells, whereas B-type lamins, arising from two different genes, are ubiquitous.
Recent familial genetic studies have shown, contrary to all expectations, that naturally occurring mutations in LMNA are responsible for two groups of diseases, apparently unrelated, affecting highly specialized tissues: dystrophies of skeletal and/or cardiac muscles, and partial lipodystrophies.
This review will first focus on the clinical aspects of these diseases and the phenotype-genotype correlations. We will summarize recent biological and experimental data on tissue and cellular alterations related to diverse molecular abnormalities in lamins A/C. This field of investigations provides informations of great interest for the understanding of the physiological role of lamins, and allows the exploration of new hypotheses on pathophysiological mechanisms leading to LMNA-linked diseases.
Introduction
Lamins are widely expressed proteins, but their precise physiological role remains still unknown. As other members of the intermediate filament protein family, their structure comprises a central a-helical coiled-coil rod domain flanked by globular N-terminal (head) and C-terminal (tail) domains.1 Lamins A and C are the major alternative splice products of the LMNA gene. Their expression is developmentally regulated, increasing during differentiation. Two genes give rise to B-type lamins, which are constitutively expressed. Both types of lamins, dephosphorylated at the end of the mitosis, polymerize through coiled-coil interactions between their a-helical rod-domains, to form the nuclear lamina at the nucleoplasmic side of the inner nuclear membrane in interphase cells. The lamina interacts with integral proteins of the inner nuclear membrane like emerin, which binds A-type lamins. In addition, the lamina has narrow intrications with chromatin and nuclear pores complexes which mediate molecular trafficking between cytoplasm and the nucleus.2
The first evidence that LMNA was linked to a genetic disease was reported in 1999. By a positional approach, studies of familial genetic linkage in a large French pedigree with autosomal dominant Emery-Dreifuss muscular dystrophy (AD-EDMD) showed a strongly positive lod-score at an 8-cM locus on chromosome 1q21-q23. LMNA, which is located in this interval, was subsequently shown to be mutated in all affected subjects.3 After this first publication, LMNA heterozygous alterations were shown to be responsible of two other diseases, which share with AD-EDMD several clinical symptoms: dilated cardiomyopathy with conduction defects (DCM-CD),4,5 and limb-girdle muscular dystrophy with cardiac conduction disturbances (LGMD1B).6
A genetic linkage between the familial partial lipodystrophy of the Dunnigan type (FPLD) and a chromosome 1q21–22 locus has been known since 1998.7–9 The discovery of LMNA mutations in this disease was surprising, since this disorder was clinically not at all suspected to be linked to muscular dystrophies.10,11
After a description of the clinical and genetic features of these two groups of diseases, we will report the recent findings on the nuclear alterations found in cells from patients and in experimental models (e.g., transgenic mice or genetically modified cell lineages). We will discuss rising pathophysiological hypotheses on these LMNA-linked diseases.
Disorders of Cardiac and/or Skeletal Muscles Linked to LMNA Alterations
Emery-Dreifuss Muscular Dystrophy (EDMD)
Benign forms of Duchenne muscular dystrophies were reported for the first time by Becker and Kiener in 1955.12 In 1961, Dreifuss and Hogan described a Virginian family with an X-linked muscular dystrophy that was considered at that time as a benign form of Duchenne muscular dystrophy (DMD).13 However, after a detailed clinical characterization of the family, Emery and Dreifuss suggested that this family presented a muscular dystrophy different from Duchenne and Becker muscular dystrophy.14 This new clinical entity was later referred to as Emery-Dreifuss muscular dystrophy (EDMD).15 Nevertheless, EDMD was most probably described for the very first time in 1902 by Cestan and Lejonne.16These authors reported a case of two brothers with a familial myopathy with severe and generalized contractures.
EDMD is a clinically and genetically heterogeneous condition. It is typically characterized by a triad of: 1) early contractures of the Achilles tendons, elbows and post-cervical muscles, often before there is any significant weakness, 2) slowly progressive muscle wasting and weakness with a distinctive humero-peroneal distribution early in the course of the disease and 3) by adult life, cardiomyopathy usually presenting as cardiac conduction defects (ranging from sinus bradycardia, prolongation of the PR interval on ECG to complete heart block requiring pacing). Thus affected individuals may die suddenly from heart block, or develop progressive heart failure. The latter may occur subsequent to the insertion of a pacemaker to correct an arrhythmia (Fig. 1, Table 1).5,17,18 Skeletal muscle biopsies from patients with EDMD show dystrophic changes with a few necrotic and regenerating fibers, but this is not specific to this muscular disease. Usually fiber necrosis is less prominent than in DMD or Becker muscular dystrophy (BMD).19 Skeletal muscles also show marked variations in fiber diameter and an increased number of hypertrophic fibers and internal nuclei.20,21
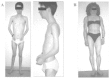
Two major modes of inheritance of EDMD exist, X-linked (XL-EDMD) and autosomal dominant (AD-EDMD). Rare cases of autosomal recessive transmission (AR-EDMD) have been also reported.22–24Defects in the emerin protein are responsible of XL-EDMD,25 whereas mutations in lamin A/C gene cause AD and AR-EDMD.3,24 The three forms are overall clinically identical,18,26,27even if some slight differences emerge between XL and AD forms of EDMD.28AD-EDMD exhibits wider clinical variability than XL-EDMD. AD-EDMD patients have more severe and progressive wasting of the biceps brachii compared to what is typically found in XL-EDMD.29,30 Hypertrophy of the quadriceps and of the extensor digitorum brevis occurs in several AD-EDMD patients but not in XL-EDMD. Contractures, which are the first symptoms in XL-EDMD, might appear after weakness and difficulty in running in AD-EDMD patients. Loss of ambulation due to a combination of increasing joint stiffness and weakness is observed in AD-EDMD but is extremely rare in XL-EDMD.18,28,31
Limb-Girdle Muscular Dystrophy Associated with Atrioventricular Conduction Disturbances (LGMD1B)
Among the large family of muscular dystrophies, “limb-girdle muscular dystrophies”(LGMD) represent a genetically heterogeneous group of myogenic disorders with a limb girdle distribution of weakness.32The inheritance pattern in LGMD is heterogeneous. Four dominant (LGMD1) and eight recessive forms (LGMD2) have been identified to date. Van der Kooi et al33 have described the LGMD1B form, inherited as an autosomal dominant trait. It is characterized by symmetrical weakness starting in the proximal lower limb muscles, and gradually proximal upper limb muscles also become affected. At variance with EDMD, early contractures of the spine are absent, and contractures of elbows or Achilles tendons are either minimal or late. Cardiac deficiency is not a constant feature in LGMD. However in LGMD1B, cardiological abnormalities are found in the majority of patients. These include dysrhythmias and atrioventricular conduction disturbances, such as bradycardia and syncopal attacks that require pacemaker implantation, and sudden cardiac death. In the three families described by van der Kooi et al,33 there was a significant relation between the severity of atrioventricular conduction disturbances and age, and neuromuscular symptoms preceded cardiological involvement. These clinical features are very close to those of EDMD, however, the late appearance or absence of contractures led the authors to conclude that this disorder differed from EDMD. Muscle biopsies from patients with LGMD1B show non-specific myopathic changes similar to those observed in XL and AD-EDMD (Table 1).33
Because the locus of LGMD1B had been mapped to chromosome 1q11–21,34where LMNA is located, the LMNA gene became a good candidate for this muscular disease. Mutation analysis of LMNA in the three LGMD1B families described by van der Kooi et al identified three different LMNA mutations. This demonstrated that LGMD1B and AD-EDMD are allelic disorders.6
Dilated Cardiomyopathy and Conduction Defects (DCM-CD)
Cardiomyopathies are defined as diseases of the myocardium associated with cardiac dysfunction.35 The most common forms are the dilated forms, responsible for approximately 60% of cases of cardiomyopathy with an annual incidence estimated to be 5–8 cases per 100,000 people.36 Dilated cardiomyopathy (DCM) is characterized by dilatation and impaired contraction of the left ventricle or both ventricles. Histology is non-specific. Presentation is usually with heart failure, which is often progressive. Arrhythmias, thromboembolism and sudden death are common and may occur at any stage. Many causes of DCM have been described, but most commonly this disease is considered idiopathic. In the past years it has become increasingly clear that in at least 25% of DCM cases have a genetic basis.37,38 Familial DCM is a heterogeneous disorder with different inheritance patterns. Autosomal dominant inheritance is the most common, but autosomal recessive, X-linked and mitochondrial inheritance have also been identified. In the last couple of years important progress has been made in unraveling familial DCM. Multiple genetic loci and several genes have been identified in this disease.
That LMNA could also be involved in DCM was completely unexpected. In the majority of affected members of one of the French families with AD-EDMD, the disease was confined exclusively to the heart and associated with arrhythmias, left ventricular dysfunction, dilated cardiomyopathy and a high incidence of sudden death.3,5 These patients with exclusive cardiac involvement could easily have been diagnosed as DCM with conduction defects (DCM-CD, Table 1), suggesting that LMNA was one of the disease genes of familial DCM-CD. In agreement with this finding, mutations in LMNA were found in unrelated families with DCM-CD.4 In addition, a LMNA mutation was identified in a family in which three phenotypes were described, DCM with EDMD-like skeletal muscle abnormalities, DCM with LGMD-like skeletal muscle abnormalities and pure DCM-CD.39 So far, no sign of lipodystrophy has been reported in patients with striated muscle disorders due to LMNA mutations.
There is no specific treatment for these cardiac and/or skeletal laminopathies. However, great care should be given to proper diagnosis and follow-up of patients with EDMD, LGMD1B or DCM-CD. All patients should have a detailed cardiac examination and regular follow-up by a cardiologist since sudden death can occur, and early detection of arrhythmias can be lifesaving by defibrillator implantation. Also relatives of such patients should be cardiologically screened even if they show no subjective neuromuscular or cardiac symptoms. Apart from the patients with a typical presentation of weakness, contractures, and arrhythmia, especially in familial cases with a history of sudden death, LMNA mutations ought to be searched for.
Spectrum of LMNA Mutations in Cardiac and/or Skeletal Muscle Disorders
Lamins A and C (664 and 572 amino acids, respectively) are encoded by the LMNA gene through alternative splicing within exon 10. LMNA contains twelve exons, which are spread over approximately 24 kb of genomic DNA (Fig. 2).40 Since the first description of LMNA mutations in AD-EDMD, a total of fifty-one different mutations were reported, forty-four in striated muscles disorders and seven in partial lipodystrophies. These mutations are: 2 nonsense mutations, 2 deletions with frameshift, 1 splice site mutation, 4 in-frame deletions and 42 missense mutations (Fig. 2). Two “hot spot” mutations are observed: the R453W missense mutation24,28,41,42in AD-EDMD and the missense mutation at R482. The R482 mutation exists in three different forms (R482W, R482Q and R482L) in FPLD.10,11,43,44
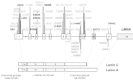
In cardiac and skeletal muscle disorders, the mutations are distributed along the gene between exons 1 and 10 in the region common to lamins A and C except for three missense mutations. One is located in exon 10 specific of lamin C and the other two in exon 11 specific of lamin A.5,41,45Thirty-five mutations were identified in 19 AD-EDMD families, 36 sporadic EDMD cases and 4 LGMD1B families.3,20,4,10,23,7,37The large proportion of EDMD sporadic cases reported underlines the high frequency of de novo mutations in the LMNA gene.28 However, if mutations in LMNA gene are identified in 100% of EDMD familial cases, they are found in only 35% of sporadic cases with EDMD-like phenotype (Bonne G., personal communication). The clinical picture is often compatible with other muscular dystrophies, explaining the low efficiency of LMNA mutation detection in the EDMD-like isolated cases.
One mutation, H222Y, was identified at a homozygous state in a patient from a consanguineous family. The unaffected parents carried the mutation at a heterozygous state, demonstrating that LMNA mutations are also responsible of AR-EDMD.24 The same amino acid, H222, was mutated to a proline (H222P) in another EDMD family with autosomal dominant transmission.28 Thus, the pathogenic effect of mutations affecting the same amino acid is variable depending on the type of amino acid change (tyrosine versus proline). Another recessive case was recently described in which two heterozygous mutations were identified, one being specific of lamin A.41 Finally, the R336Q mutation was identified in an EDMD family with an autosomal dominant transmission in which only two out of the four members carrying the mutation were affected.24Altogether, theses results demonstrate that LMNA mutations have variable pathogenic effects with a semi-dominant, dominant or recessive pattern of expression.24,41
Eleven mutations have been reported in patients with isolated DCM.4,5,39,45,48 It was initially suggested that mutations in either the rod domain or the tail domain of lamin C might underlie the DMC-CD phenotype.4 However, two reports demonstrated that within a family, the same LMNA mutation gives rise to various phenotypes ranging from an isolated DCM to LGMD1B or EDMD.5,39
Among all the reported cases with cardiac and/or skeletal disorders (EDMD, LGMD1B and DCM) due to LMNA mutation, there is no clear correlation between the phenotype and type or localization of the mutation in the gene.5,6,24,28,39,47 Further studies are needed to identify the factors modifying striated muscle phenotypes among patients harboring mutations within lamin A/C. In contrast, 90% of mutations described in FPLD affect the same codon in exon 8 (arginine 482 is mutated in 28/32 reported FLPD cases). Besides this “hot spot”, four other mutations were reported in FPLD families, two located in exon 8 and two in exon 11 that encodes only the tail domain of lamin A.10,11,43,44
Lipodystrophies and the Familial Partial Lipodystrophy of the Dunnigan Type (FPLD)
Lipodystrophies
Lipodystrophies represent a heterogeneous group of diseases characterized by generalized or partial alterations in body fat development or distribution and insulin resistance. The other cardinal clinical signs of these syndromes are acanthosis nigricans, which is a skin disorder associated with insulin resistance, frequent hyperandrogenism in females, muscular hypertrophy and liver steatosis. Insulin resistance is associated with a progressive altered glucose tolerance leading to diabetes, and with hypertriglyceridemia.49,50 When chronic hyperglycemia occurs, its treatment is often very difficult due to major insulin resistance, and the condition leads to early diabetic complications. Acute pancreatitis due to severe hypertriglyceridemia, and liver cirrhosis arising from the frequent nonalcoholic steatohepatitis are also responsible for the morbidity and mortality of these diseases. However, a broad pattern of severity is seen in lipodystrophies, ranging from the rare and serious congenital generalized form to the milder acquired partial one.
The main forms of lipodystrophies are classified according to their origin, either genetic or acquired, and to the clinical pattern of the lipoatrophy, either generalized or partial (Table 2). Etiologies of lipodystrophies are very heterogeneous. Two genetic loci, on chromosomes 9q34 and 11q13 are linked to the congenital generalized lipodystrophy (Berardinelli-Seip syndrome), which is transmitted as an autosomal recessive trait.51,52 Recently, the 11q13 locus has been identified as the BSCL2 gene, encoding seipin, a protein of unknown function mainly expressed in brain.52
LMNA is presently the only gene known to be involved in a genetic syndrome of partial lipodystrophy, i.e. the familial partial lipodystrophy of the Dunnigan-type (FPLD). However, it was shown that dominant negative mutations in PPARγ, a transcription factor involved in adipogenesis, lead to severe insulin resistance, diabetes, and hypertension.53 Recent studies suggest that affected subjects also present subcutaneous paucity of fat on limbs (S. O'Rahilly, personal communication).
Some forms of lipodystrophies could have an immunological basis: autoimmune diseases are sometimes associated with sporadic cases of generalized lipoatrophies (e.g., Lawrence syndrome). The C3 nephritic factor, an IgG antibody against complement components, can be detected in some cases of partial lipodystrophy of the Barraquer-Simons type (lipoatrophy of face and trunk, with excess accumulation of fat in the lower part of the body).54 Finally, the probably most frequent form of lipodystrophy is the redistribution of fat that occurs in HIV-infected patients, mainly treated by antiretroviral medications. These patients frequently lose peripheral subcutaneous fat, accumulate visceral adipose tissue, and develop hypertriglyceridemia and insulin resistance.55
The pathophysiology of lipodystrophies is still unknown. However, murine models of lipoatrophic diabetes (aP2-nSREBP-1c and A-ZIP/F-1 mice) revealed that primary genetic alterations in fat development resulted in diabetes and dyslipidemia.56,57 Diabetes could be reversed by fat transplantation in the A-ZIP/F-1 model.58 Leptin deficiency, caused by the absence of adipose tissue, could be an important determinant of the metabolic abnormalities since exogenous administration or transgenic overexpression of leptin has been shown to markedly improve insulin sensitivity, glycemic control, dyslipidemia and hepatic steatosis in these mice.59,60 Similarly, the defect in adiponectin, another fat-derived hormone, has recently been shown to be involved in insulin resistance.61 Regarding the HIV-linked lipodystrophy syndrome, several authors recently pointed out the deleterious effects of some antiretroviral treatments on adipogenesis.62–64 Altogether, these studies provide strong arguments for a primary role of disturbances in fat distribution or development, which lead secondarily to insulin resistance and metabolic complications.
Partial lipodystrophies with insulin resistance are pathophysiological models of great interest for diabetologists. Indeed, they represent stereotyped forms of the metabolic syndrome, largely prevalent in developed countries, which associates android repartition of fat, glucose intolerance, hypertension and dyslipidemia.65 This frequent condition represents a strong risk factor for cardiovascular diseases. As abnormalities in the body distribution of fat are possible important primary etiologic factors for the development of type 2 diabetes,66,67 partial lipodystrophies constitutes a new field of investigation in the pathophysiology of diabetes.
Familial Partial Lipodystrophy of the Dunnigan Type (FPLD)
Among lipodystrophic syndromes, the familial partial lipodystrophy of the Dunnigan-type (FPLD), dominantly inherited, is a rare disease characterized by the disappearance, after puberty, of adipose tissue in the limbs, buttocks and trunk. This progressive lipoatrophy spares the neck and face, where adipose tissue can accumulates, causing frequently a cushingï appearance.68 However, in lean patients, this latter feature can be lacking, and differential diagnosis with total acquired lipoatrophy can be difficult if the familial dominant transmission of the disease is not evident. Prominence of muscles and superficial veins are partly due to the lipoatrophy, but, as in the other syndromes of insulin resistance, a genuine muscular hypertrophy is present.69 The android aspect of patients is particularly striking in females, but does not systematically draw attention in males, in which this condition is frequently unrecognized. Garg et al performed magnetic resonance imaging studies in four affected patients, three females and one male.70 They confirmed the clinically observed altered distribution of the subcutaneous adipose tissue, near-totally absent in areas from extremities and gluteal region, reduced in the truncal area, and increased in the neck and face. Intra-abdominal, intra-thoracic, and intermuscular fat is preserved, as well as mechanical adipose tissue (present in orbits, palm, sole, scalp and periarticular regions). In females, breasts have a markedly reduced subcutaneous fat whereas adipose tissue accumulates in the labia majora.70 The partial adipose tissue loss in FPLD is associated with a reduced plasma leptin level, to about 40% of normal.71
Metabolic alterations associated with FPLD are responsible for the severity of the disease. Insulin resistance, usually attested by hyperinsulinemia with concomitant normal or elevated glycemia, has been confirmed by several tests, including euglycemic-hyperinsulinemic clamp.72 Insulin-stimulated glucose transport and/or oxidation was found to be impaired in neck and abdomen adipocytes from affected patients, despite normal insulin binding, showing a post-receptor defect in the insulin action.72 Clinically, acanthosis nigricans, brownish hyperkeratotic skin affection localized to the axillary and inguinal folds, is associated with insulin resistance. When hyperinsulinemia no longer compensates for insulin resistance, glucose intolerance, then diabetes, occur.
Dyslipidemia is frequent among FPLD patients. Hypertriglyceridemia, due to elevated very light density lipoprotein (VLDL) level, is the more prevalent feature. When severe, it can lead to a life-threatening acute pancreatitis. Other perturbations of the lipid profile can also be associated with FPLD, as decreased high-density lipoprotein (HDL)-cholesterol levels, with or without elevated total cholesterol.73,74 As hypertension is also frequent in FPLD, these patients accumulate numerous cardiovascular risks, leading to early coronary heart disease.75 Like in other lipodystrophic syndromes, a liver steatosis, with its risk of evolution towards cirrhosis, usually occurs in these patients.
Although this disease affects males and females, both clinical traits and metabolic complications are more severe in women.44,76 Accordingly, this disease was first reported only in females, and a X-linked dominant transmission was initially evoked.68 In addition, women affected by FPLD frequently complain of hirsutism, and a polycystic ovary syndrome with ovarian dysfunction, and hyperandrogenism is usual. So far, no specific cardiomyopathy or skeletal muscular dystrophy linked to LMNA mutations have been reported in FPLD patients.
Treatment of FPLD is difficult. Appropriate diet and physical training are important to minimize metabolic alterations. However, diabetes mellitus, which appears secondarily in the evolution of the disease, requires usually large doses of insulin. Insulin sensitizers, like metformin, could improve the control of glycemia. PPARγ agonists, such as thiazolidinediones, which promote both adipocyte differentiation and insulin sensitivity, seem promising in lipodystrophic syndromes.77 A leptin treatment is currently evaluated in these patients.
Mutations causing FPLD cluster only in exons 8 and 11 of LMNA, coding for the globular C-terminal domain of type A-lamins (Fig. 2).10,11,43,44,78,79They all affect highly conserved residues among species. The most frequent mutation substitutes a basic amino acid at position 482 (arginine) for a neutral residue (tryptophan, glutamine, leucine). All patients are heterozygous for these mutations. The mutations occur in several different haplotypes in the families, suggesting that codon 482 is a site of recurrent mutation in unrelated pedigrees. The critical location at codon 482 of FPLD-linked LMNA alterations was recently confirmed by the observation of a patient with a LGMD1B phenotype due to a missense mutation at the adjacent codon 481.47The deamination of C to T at a CpG site is a likely mutational mechanism in the case of the R482W substitution. Other mutations also induce a complete or partial loss of a positive charge (from lysine for K486N substitution, or from arginine in the mutations R582H or R584H which affect exon 11) or the appearance of a negative charge (aspartate in the G465D mutation). Alterations in exon 11 are rare, concerning two reported families for the R584H alteration44,78 and only one for the R582H substitution.43 They affect specifically the A isoform of lamin. Two sisters affected by the R582H substitution have a less severe loss of subcutaneous adipose tissue and milder metabolic abnormalities.79 However, we did not observe such an attenuate phenotype in the patient harboring the R584H mutation that we studied.44 Environmental factors, such as diet and physical activity, are probably important determinants of the severity of metabolic complications.78
Studies of LMNA in other metabolic disorders have been performed. We excluded LMNA mutations as being responsible for generalized lipodystrophy.44No mutations in exon 8 of LMNA have been found in subjects with HIV therapy-associated lipodystrophy. Furthermore, lamins A/C and HIV-1 protease do not have any sequence homology, providing no evidence for the direct inhibition of lamins by HIV-1 protease inhibitors.80Hegele et al suggested that a common variation in LMNA could be associated with obesity-related phenotypes.81,82 However, there is presently no evidence for an association of this variant with type 2 diabetes.83
Could Some Patients with LMNA Mutations be Affected by Both Skeletal or Cardiac Muscular Symptoms and Lipodystrophy?
The association of LMNA mutation-linked skeletal or cardiac muscular defects with lipodystrophy or metabolic abnormalities has not been reported so far. A muscular hypertrophy is usual in FPLD, as in other lipodystrophic syndromes. Calf hypertrophy could be reminiscent of what is observed in LGMD1B. Although some patients complain of cramps, their muscular strength is normal in most cases. However, patients with FPLD due to the LMNA R482W mutation and presenting muscular signs compatible with LGMD1B are being investigated.84 A cardiac septal hypertrophy can be observed in FPLD patients, but is difficult to attribute to a specific genetic defect since it could be secondary to diabetes and hypertension. Further investigations of neuromuscular and cardiac phenotype in FPLD patients are needed. Likewise, an accurate evaluation of adipose tissue distribution, insulin sensitivity and lipid metabolism has not been reported in patients with AD-EDMD, LGMD1B or DCM-CD linked to LMNA mutations.
Experimental Models of Lamin A/C Alterations
lmna Knock-Out Mice
Sullivan et al reported the derivation of mice in which the lamins A/C have been eliminated by gene targeting (by deletion of a region extended from exon 8 to the middle of exon 11), to produce either homozygous or heterozygous offspring.85 Homozygous mice, although normal at birth, rapidly exhibit a striking cardiac and skeletal muscular dystrophy with rigidity, more marked in proximal muscles, that resembles the EDMD phenotype in humans. In addition, postnatal growth is severely impaired and the mice exhibit premature mortality. Although a white fat atrophy has also been reported in this model, additional metabolic features of a lipodystrophic syndrome have not been described, so this appearance could be related to cachexia. Mice heterozygous for the lmna mutation are overtly normal at 6–10 months with minimal evidence of dystrophy.
Studies of cells from these mice clearly showed that loss of lamin A/C affects nuclear envelope integrity.85Indeed, nuclei from embryonic lmna-/- fibroblasts often exhibited an abnormal shape, with nuclear regions displaying disruption of heterochromatin, withdrawal of B-type lamins and other proteins of the inner nuclear envelope such as lamina-associated polypeptide (LAP)2b, irregular distribution of nuclear pores complexes, and partial mislocalization of emerin to the cytoplasm. Interestingly, this aberrant emerin distribution was rescued by overexpression of wild-type or R482W-mutated lamin A, contrary to the L85R, N195K or L530P mutated forms of the protein, which had no effect on emerin relocalization.85,86
Nuclear Alterations in Cells Harboring LMNA Mutations
From the studies in lmna-/- mice, it could be postulated that a loss of lamin A/C protein expression or function could underlie the muscular phenotypes. One the other hand, from studies of patients with XL-EDMD, it appears that the disease arises from either loss of emerin protein or mutations resulting in its subcellular mislocalization.87–89 XL and AD-EDMD are almost clinically indistinguishable, thus a pathophysiological hypothesis would be that a defect in emerin distribution, secondary to LMNA mutations, is responsible for EDMD, as also suggested by studies of lmna-/- cells. However, heterozygous lmna+/- mice are healthy, and although their cells present frequent irregular nuclei, nuclear envelope proteins show a largely normal distribution.85
In human, immunocytochemical analysis of lamin A/C and emerin on skeletal muscle biopsies of AD-EDMD patients carrying LMNA mutation showed no detectable differences from control muscles, indicating that the mutations do not significantly alter the structure of the nuclear envelope.21 The same analysis performed on a cardiac biopsy of an AD-EDMD patient with a non-sense mutation showed the same results.3 The latter mutation leads potentially to the production of truncated lamins A/C with only five amino acids. Most probably such small peptides are degraded and only lamins A/C produced by the intact allele are expressed in the patients. This hypothesis was confirmed by the Western-blot analysis of the explanted myocardial tissue that showed a decreased expression of lamins A/C compared to that of a control heart tissue.5
In some of EDMD patients, an ultrastructural examination of the skeletal muscle biopsies showed a loss of heterochromatin from wide stretches along the nuclear envelope, in 10% of nuclei, with a rarefaction of nuclear pores complexes in these areas. Irregular shapes of some nuclei are also reported.21,90However, only a small proportion of muscle nuclei exhibit abnormalities, thus study of additional cases is required to draw any conclusion. Nevertheless, these alterations are reminiscent of features of lmna-/- mouse cells, albeit affecting a lower percentage of cells, and in skeletal muscle of XL-EDMD patients.91,92 Further work is required using cells from patients with skeletal or cardiac muscular disease linked to LMNA alterations in order to evaluate the extent of nuclear disturbances.
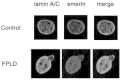
We recently performed a study of skin fibroblasts of FPLD patients with R482W and R482Q LMNA mutations.93 Protein expression of type A and type B lamins, LAP2b and emerin was normal in the whole population of fibroblasts. However, 5–25% of these cells had abnormal blebbing nuclei with A-type lamins forming a peripheral meshwork that was frequently disorganized (Fig. 3). Emerin strictly colocalized with this abnormal lamin A/C meshwork (Fig. 4), in agreement with the R482W mutated-lamin A dependent relocalization of emerin in lmna-/- fibroblasts,86 and the preserved interactions between R482Q lamin A and emerin in vitro.94Cells from lipodystrophic patients often had other nuclear envelope defects, mainly herniations deficient in B-type lamins, nuclear pores and LAP2b. Furthermore, heterogeneous DNA staining by DAPI suggested that chromatin was decondensed in nuclear areas flanking disorganized nuclear envelope domains (Fig. 3). The mechanical properties of nuclear envelopes were altered, as judged from the extensive deformations observed in nuclei from heat-shocked cells, and from the low stringency of extraction of nuclear envelope proteins. These structural nuclear alterations were caused by the lamins A/C mutations, since the same changes were introduced in human control fibroblasts by expression of R482W mutated lamin A. However, despite these abnormalities, we showed that the fibroblasts from FPLD patients were euploid and able to cycle and divide.

Recently, transfection studies of mutant LMNA alleles have been performed in several cell types by three groups (Favreau C, Östlund C, Worman HJ, Courvalin JC, Buendia B, personal communication).86,95 Data in HeLa cells and C2C12 myoblasts show heterogeneous defects in the shape of the nuclei, in the assembly of type A-lamins and/or in the distribution of emerin that concern some but not all EDMD or DCM-linked LMNA mutations. In human fibroblasts and in C2C12 myoblasts, overexpression of lamin A mutated in the carboxy-terminal domain generated an aberrant nuclear phenotype similar to that observed in cells from FPLD patients (Favreau C, Östlund C, Worman HJ, Courvalin JC, Buendia B, personal communication).93
The observation of heterogeneous, but very similar nuclear alterations in fibroblasts from FPLD patients or from XL-EDMD patients, from lmna-/- mice and from myocytes of AD-EDMD patients confirms that lamins A/C are major determinants of the nuclear architecture. However, these studies have shown that A-type lamins are not the only determinants of emerin localization. If altered interactions between FPLD-linked mutant forms of lamin A/C and emerin do not seem likely in FPLD, they probably neither represent the only pathophysiological mechanism in cardiac and skeletal muscular dystrophies linked to LMNA mutations.
Conclusion
Recent experiments evidenced the essential role of A-type lamins in nuclear architecture and integrity. However, the pathophysiology of the diseases linked to LMNA mutations remains unclear. A striking feature of these diseases is the tissue-specificity of the alterations, which is difficult to relate to the widespread expression of lamin A/C in differentiated cells.
Germline mutations of the RET protooncogene is another example of different phenotypes arising from mutations in the same gene. However, in the case of RET mutations, the phenotypes of either to congenital malformations or inherited cancer syndromes are due to the loss or gain of function of the mutated protein.96 The divergent consequences of alterations in lamin A/C, a structural protein, seem more complex to understand. Knowledge about the other specific functions of lamin A/C is presently lacking to unravel the pathophysiological mechanisms of muscular dystrophies and lipodystrophies linked to LMNA. However, several hypotheses can be discussed.
The diverse types of LMNA mutations in patients with EDMD, LGMD1B and DCM-DC, some of them leading to a truncated form of the protein, suggest that a loss of function of lamin A/C could be responsible for the cardiac and/or skeletal muscle diseases. This loss of function could act via a secondary mislocalization of emerin, as suggested by the close clinical symptomatology induced by emerin mutations. However, from present cellular studies, emerin alterations were not systematically found when the LMNA gene was mutated, suggesting that this mechanism might not be unique. Further work on cells from patients will most likely provide more detailed information on this issue.
The compromised nuclear integrity could lead to nuclear fragility, and to mechanical damage during muscle contractions that could explain the striated muscle affections. However, the preferential alteration of the conduction pathway in cardiac tissue in LMNA-linked muscular dystrophies and cardiomyopathies and the nuclear fragility that we recently evidenced in cells from FPLD patients with LMNA R482W or R482Q mutations, are not in favor of this hypothesis.93
The mutational hot-spot found in FPLD-linked phenotype suggests a more specific alteration of a lamin A/C function that could only be expressed at the adipocyte level. The systematic alteration of the charge of an amino acid in the C terminal domain of the protein could evoke modifications of the binding of lamin A/C with others partners. The recent crystalization of the C-terminal end of lamin A showed that it is spatially organized as an immunoglobulin-like domain and that mutations in amino acids at positions 465, 482 and 486, found in FPLD, are localized at the external surface of the structure (S. Dhe-Paganon, E. Warner, S. Schoelson, personal communication). As immunoglobulin folds are known to mediate protein-protein, protein-lipid and protein-DNA interactions, this hypothesis could have a pathophysiological relevance, but data about of lamin A/C partners inducing a tissue-specific response are presently lacking.
A role for the inner nuclear membrane in the regulation of gene expression has been previously suggested.97 In yeast, the inner nuclear membrane is involved in the spatial organization of chromatin and in the regulation of transcription.98In mammalian species, gene expression is also influenced by the spatial organization of the nucleus (for a review, see ref. 99). Disturbances in heterochromatin organization are a largely represented feature in cells affected by LMNA mutations. It is tempting to speculate that lamins could alter tissue-specific gene expression, and that the mutations could affect muscle or adipose tissue function, differentiation or survival. Indeed, several transcription factors, among which the retinoblastoma protein pRb, a key regulator of cell-cycle dependent transcription, are known to interact with A type-lamins (for a review, see refs. 100–102. Furthermore, the lamina provides an attachment site for apoptotic signaling machinery100and nuclear envelope proteins are early targets for caspase degradation.103
Finally, it has recently been shown in Drosophila that lamins, in addition to their role in nucleus integrity, are also required for cytoplasmic organization,104 in accordance with the known mechanical interactions between nuclear scaffolding proteins and cytoskeletal filaments.105 This aspect has now to be investigated in mammalian LMNA-mutated cells. Constant improvements in the description of lamin A/C-associated diseases and in experimental models for alterations in nuclear envelope proteins will provide important information for the understanding of nuclear physiology, adipose tissue and metabolic diseases, and skeletal and cardiac muscle dystrophies.
Addendum
During the editing process of this communication, a new phenotype also due to mutation of lamin A/C gene has been reported. This fifth laminopathy disease is an autosomal recessive form of axonal neuropathy (Charcot-Marie-Tooth disorder type 2, or CMT2B1).106 The Charcot-Marie-Tooth (CMT) disorders comprise a group of clinically and genetically heterogeneous hereditary motor and sensory neuropathies, which are mainly characterized by muscle weakness and wasting, foot deformations, and electrophysiological as well as histological changes. A subtype, CMT2, is defined by a slight or absent reduction of nerve-conduction velocities together with the loss of large myelinated fibers and axonal degeneration. Homozygosity mapping in inbred Algerian families with autosomal recessive CMT2 (AR-CMT2) provided evidence of linkage to chromosome 1q21.2-q21.3 in two families. All patients shared a common homozygous ancestral haplotype that was suggestive of a founder mutation as the cause of the phenotype. A unique homozygous mutation in LMNA was identified in all affected members and in additional patients with CMT2 from a third, unrelated family. Ultrastructural exploration of sciatic nerves of LMNA null (i.e., LMNA/-) mice was performed and revealed a strong reduction of axon density, axonal enlargement, and the presence of non-myelinated axons, all of which were highly similar to the phenotypes of human peripheral axonopathies. The association of nerve abnormality with LMNA mutations enlarges the already broad range of phenotypes of laminopathies and shed further light on the important interactions between nerve and striated muscle.
Acknowledgments
Figure 1A is a courtesy of Prof J-A. Urtizberea, Service de Mèdecine Physique et Rèadaptation de l'Enfant, Hôpital Raymond Poincarè, Garches, France. Figure 2 was adapted with permission from Roberts and Schwartz, 2000.107 The authors thank Brigitte Buendia, Jean-Claude Courvalin and Jacqueline Capeau for critical reading of the manuscript.
References
- 1.
- Stuurman N, Heins S, Aebi U. Nuclear lamins: Their structure, assembly, and interactions. J Struct Biol. 1998;122:42–66. [PubMed: 9724605]
- 2.
- Worman HJ, Courvalin JC. The inner nuclear membrane. J Membr Biol. 2000;177:1–11. [PubMed: 10960149]
- 3.
- Bonne G, Di Barletta MR, Varnous S. et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nature Genet. 1999;21:285–288. [PubMed: 10080180]
- 4.
- Fatkin D, MacRae C, Sasaki T. et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. [PubMed: 10580070]
- 5.
- Bécane H -M, Bonne G, Varnous S. et al. High incidence of sudden death of conduction system and myocardial disease due to lamins A/C gene mutation. Pacing Clin Electrophysiol. 2000;23:1661–1666. [PubMed: 11138304]
- 6.
- Muchir A, Bonne G, van der Kooi AJ. et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000;9:1453–1459. [PubMed: 10814726]
- 7.
- Peters J, Barnes R, Bennett L. et al. Localization of the gene for familial lipodystrophy (Dunnigan variety) to chromosome 1q21–22. Nat Genet. 1998;18:292–295. [PubMed: 9500556]
- 8.
- Jackson SN, Pinkney J, Bargiotta A. et al. A defect in the regional deposition of adipose tissue (partial lipodystrophy) is encoded by a gene at chromosome 1q. Am J Hum Genet. 1998;63:534–540. [PMC free article: PMC1377312] [PubMed: 9683602]
- 9.
- Anderson JL, Khan M, David WS. et al. Confirmation of linkage of hereditary partial lipodystrophy to chromosome 1q21–22. Am J Med Genet. 1999;82:161–165. [PubMed: 9934982]
- 10.
- Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109–112. [PubMed: 10587585]
- 11.
- Shackleton S, Lloyd DJ, Jackson SN. et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24:153–156. [PubMed: 10655060]
- 12.
- Becker PE, Kiener F. Eine neue X-chromosomale muskeldystrophie. Arch Psychiatr Nervenkr. 1955;193:427. [PubMed: 13249581]
- 13.
- Dreifuss FE, Hogan GR. Survival in X-chromosomal muscular dystrophy. Neurology. 1961;11:734–737. [PubMed: 13724309]
- 14.
- Emery A E H, Dreifuss FE. Unusual type of benign X-linked muscular dystrophy. J Neurol Neurosurg Psychiat. 1966;29:338–342. [PMC free article: PMC1064196] [PubMed: 5969090]
- 15.
- Rowland LP, Fetell M, Olarte M. et al. Emery-Dreifuss muscular dystrophy. Ann Neurol. 1979;5:111–117. [PubMed: 426473]
- 16.
- Cestan R, LeJonne Une myopathie avec rétractions familiales. Nouvelle iconographie de la Salpètrière. 1902;15:38–52.
- 17.
- Emery A E H. Emery-Dreifuss muscular dystrophyA 40 year retrospective. Neuromusc Disord. 2000;10:228–232. [PubMed: 10838246]
- 18.
- Wehnert M, Muntoni F. 60th ENMC International Workshop: Non X-linked Emery-Dreifuss Muscular Dystrophy, 5-7 June 1998. Neuromusc Disord. 1999;9:115–120. [PubMed: 10220867]
- 19.
- Hopkins LC, Warren S. Emery-Dreifuss muscular dystrophy In: Rowland LP, DiMauro S, eds.Handbook of Clinical Neurology: Myopathies 18 Amsterdam: Elsevier Science,1992145–160.
- 20.
- Hausmanowa-Petrusewicz I. The Emery-Dreifuss disease. Neuropathol Pol. 1988;26:265–281. [PubMed: 3253606]
- 21.
- Sewry CA, Brown SC, Mercuri E. et al. Skeletal muscle pathology in autosomal dominant Emery-Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol Appl Neurobiol. 2001; 27:281–290. [PubMed: 11532159]
- 22.
- Takamoto K, Hirose K, Uono M. et al. A genetic variant of Emery-Dreifuss disease. Muscular dystrophy with humeropelvic distribution, early joint contracture, and permanent atrial paralysis. Arch Neurol. 1984;41:1292–1293. [PubMed: 6497732]
- 23.
- Taylor J, Sewry CA, Dubowitz V. et al. Early onset, autosomal recessive muscular dystrophy with Emery-Dreifuss phenotype and normal emerin expression. Neurology. 1998;51:1116–1120. [PubMed: 9781539]
- 24.
- di Barletta MR, Ricci E, Galluzzi G. et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2000;66:1407–1412. [PMC free article: PMC1288205] [PubMed: 10739764]
- 25.
- Bione S, Maestrini E, Rivella S. et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. [PubMed: 7894480]
- 26.
- Fenichel GM, Sul YC, Kilroy AW. et al. An autosomal-dominant dystrophy with humeropelvic distribution and cardiomyopathy. Neurology. 1982;32:1399–1401. [PubMed: 6890649]
- 27.
- Miller RG, Layzer RB, Mellenthin MA. et al. Emery-Dreifuss muscular dystrophy with autosomal dominant transmission. Neurology. 1985;35:1230–1233. [PubMed: 4022362]
- 28.
- Bonne G, Mercuri E, Muchir A. et al. Clinical and molecular genetic spectrum of autosomal dominant Emery Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48:170–180. [PubMed: 10939567]
- 29.
- Emery A E H. X-linked muscular dystrophy with early contractures and cardiomyopathy (Emery-Dreifuss type). Clin Genet. 1987;32:360–367. [PubMed: 3319295]
- 30.
- Yates JR. 43rd ENMC International Workshop on Emery-Dreifuss Muscular Dystrophy, 22 June 1996, Naarden, The Netherlands. Neuromuscul Disord. 1997;7:67–69. [PubMed: 9132143]
- 31.
- Hoeltzenbein M, Karow T, Zeller JA. et al. Severe clinical expression in X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 1999;9:166–170. [PubMed: 10382910]
- 32.
- Bushby KM. The limb-girdle muscular dystrophies-multiple genes, multiple mechanisms. Hum Mol Genet. 1999;8:1875–1882. [PubMed: 10469840]
- 33.
- van der Kooi AJ, Ledderhof TM, de Voogt WG. et al. A newly recognized autosomal dominant limb girdle muscular dystrophy with cardiac involvement. Ann Neurol. 1996;39:636–642. [PubMed: 8619549]
- 34.
- van der Kooi AJ, van Meegen M, Ledderhof TM. et al. Genetic localization of a newly recognized autosomal dominant limb-girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11–21. Am J Hum Genet. 1997;60:891–895. [PMC free article: PMC1712459] [PubMed: 9106535]
- 35.
- Richardson P, McKenna W, Bristow M. et al. Report of the 1995 World Health Organisation/International Society and Federation of Cardiology task force on the definition and classification of cardiomyopathies. Circulation. 1995;93:841–842. [PubMed: 8598070]
- 36.
- Codd MB, Sugrue DD, Gersh BJ. et al. Epidemiology of idiopathic dilated and hypertrohic cardiomyopathy. Circulation. 1989;80:564–572. [PubMed: 2766509]
- 37.
- Michels VV, Moll PP, Miller FA. et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. [PubMed: 1727235]
- 38.
- Keeling PJ, Gang Y, Smith G. et al. Familial dilated cardiomyopathy in the United Kingdom. Br Heart J. 1995;73:417–421. [PMC free article: PMC483856] [PubMed: 7786655]
- 39.
- Brodsky GL, Muntoni F, Miocic S. et al. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation. 2000;101:473–476. [PubMed: 10662742]
- 40.
- Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993; 268:16321–16326. [PubMed: 8344919]
- 41.
- Brown CA, Lanning RW, McKinney KQ. et al. Novel and recurrent mutations in lamin A/C in patients with Emery- Dreifuss muscular dystrophy. Am J Med Genet. 2001;102:359–367. [PubMed: 11503164]
- 42.
- Colomer J, Iturriaga C, Bonne G. et al. Autosomal dominant Emery-Dreifuss muscular dystrophy: : a new family with late diagnosis. Neuromusc Disord. 2002;12:19–25. [PubMed: 11731280]
- 43.
- Speckman RA, Garg A, Du F. et al. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet. 2000;66:1192–1198. [PMC free article: PMC1288186] [PubMed: 10739751]
- 44.
- Vigouroux C, Magré J, Vantyghem MC. et al. Lamin A/C gene: Sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes. 2000;49:1958–1962. [PubMed: 11078466]
- 45.
- Genschel J, Bochow B, Kuepferling S. et al. A R644C mutation within lamin A extends the mutations causing dilated cardiomyopathy. Hum Mutat. 2001;17:154. [PubMed: 11180602]
- 46.
- Felice KJ, Schwartz RC, Brown CA. et al. Autosomal dominant Emery-Dreifuss dystrophy due to mutations in rod domain of the lamin A/C gene. Neurology. 2000;55:275–280. [PubMed: 10908904]
- 47.
- Kitaguchi T, Matsubara S, Sato M. et al. A missense mutation in the exon 8 of lamin A/C gene in a Japanese case of autosomal dominant limb-girdle muscular dystrophy and cardiac conduction block. Neuromusc Disord. 2001;11:542–546. [PubMed: 11525883]
- 48.
- Jakobs PM, Hanson EL, Crispell KA. et al. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J Card Fail. 2001;7:249–256. [PubMed: 11561226]
- 49.
- Moller DE, O'Rahilly S. Syndromes of severe insulin resistance: clinical and patho-physiological features In: Moller DE, ed.Insulin resistance New York: Wiley and Sons,199349–81.
- 50.
- Reitman ML, Arioglu E, Gavrilova O. et al. Lipoatrophy revisited. Trends Endocrinol Metab. 2000;11:410–416. [PubMed: 11091118]
- 51.
- Garg A, Wilson R, Barnes R. et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84:3390–3394. [PubMed: 10487716]
- 52.
- Magré J, Delépine M, Khallouf E. et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28:365–370. [PubMed: 11479539]
- 53.
- Barroso I, Gurnell M, Crowley VE. et al. Dominant negative mutations in human PPAR-gamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–883. [PubMed: 10622252]
- 54.
- Levy Y, George J, Yona E. et al. Partial lipodystrophy, mesangiocapillary glomerulonephritis, and complement dysregulation. An autoimmune phenomenon. Immunol Res. 1998;18:55–60. [PubMed: 9724849]
- 55.
- Carr A, Samaras K, Burton S. et al. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12:F51–F58. [PubMed: 9619798]
- 56.
- Moitra J, Mason MM, Olive M. et al. Life without white fat: a transgenic mouse. Genes Dev. 1998;12:3168–3181. [PMC free article: PMC317213] [PubMed: 9784492]
- 57.
- Shimomura I, Hammer RE, Richardson JA. et al. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: Model for congenital generalized lipodystrophy. Genes Dev. 1998;12:3182–3194. [PMC free article: PMC317215] [PubMed: 9784493]
- 58.
- Gavrilova O, Marcus-Samuels B, Graham D. et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105:271–278. [PMC free article: PMC377444] [PubMed: 10675352]
- 59.
- Shimomura I, Hammer RE, Ikemoto S. et al. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–76. [PubMed: 10485707]
- 60.
- Ebihara K, Ogawa Y, Masuzaki H. et al. Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes. 2001;50:1440–1448. [PubMed: 11375346]
- 61.
- Yamauchi T, Kamon J, Waki H. et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. [PubMed: 11479627]
- 62.
- Zhang B, MacNaul K, Szalkowski D. et al. Inhibition of adipocyte differentiation by HIV protease inhibitors. J Clin Endocrinol Metab. 1999;84:4274–4277. [PubMed: 10566684]
- 63.
- Dowell P, Flexner C, Kwiterovich PO. et al. Suppression of preadipocyte differentiation and promotion of adipocyte death by HIV protease inhibitors. J Biol Chem. 2000;275:41325–41332. [PubMed: 11018036]
- 64.
- Caron M, Auclair M, Vigouroux C. et al. The HIV protease inhibitor indinavir impairs sterol regulatory element-binding protein-1 intranuclear localization, inhibits preadipocyte differentiation, and induces insulin resistance. Diabetes. 2001; 50:1378–1388. [PubMed: 11375339]
- 65.
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. [PubMed: 3056758]
- 66.
- Danforth E Jr. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26:13. [PubMed: 10973236]
- 67.
- Joffe BI, Panz VR, Raal FJ. From lipodystrophy syndromes to diabetes mellitus. Lancet. 2001;357:1379–1381. [PubMed: 11356429]
- 68.
- Köbberling J, Dunnigan MG. Familial partial lipodystrophy: two types of an X linked dominant syndrome, lethal in the hemizygous state. J Med Genet. 1986;23:120–127. [PMC free article: PMC1049565] [PubMed: 3712389]
- 69.
- Wildermuth S, Spranger S, Spranger M. et al. Köbberling-Dunnigan syndrome: A rare cause of generalized muscular hypertrophy. Muscle Nerve. 1996;19:843–847. [PubMed: 8965837]
- 70.
- Garg A, Peshock RM, Fleckenstein JL. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab. 1999;84:170–174. [PubMed: 9920078]
- 71.
- Hegele RA, Cao H, Huff MW. et al. LMNA R482Q mutation in partial lipodystrophy associated with reduced plasma leptin concentration. J Clin Endocrinol Metab. 2000;85:3089–3093. [PubMed: 10999791]
- 72.
- Ursich MJ, Fukui RT, Galvao MS. et al. Insulin resistance in limb and trunk partial lipodystrophy (type 2 Köbberling-Dunnigan syndrome). Metabolism. 1997;46:159–163. [PubMed: 9030822]
- 73.
- Hegele RA, Anderson CM, Wang J. et al. Association between nuclear lamin A/C R482Q mutation and partial lipodystrophy with hyperinsulinemia, dyslipidemia, hypertension, and diabetes. Genome Res. 2000;10:652–658. [PMC free article: PMC310873] [PubMed: 10810087]
- 74.
- Schmidt HH, Genschel J, Baier P. et al. Dyslipemia in familial partial lipodystrophy caused by an R482W mutation in the LMNA gene. J Clin Endocrinol Metab. 2001;86:2289–2295. [PubMed: 11344241]
- 75.
- Hegele RA. Premature atherosclerosis associated with monogenic insulin resistance. Circulation. 2001;103:2225–2229. [PubMed: 11342468]
- 76.
- Garg A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab. 2000;85:1776–1782. [PubMed: 10843151]
- 77.
- Arioglu E, Duncan-Morin J, Sebring N. et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med. 2000;133:263–274. [PubMed: 10929166]
- 78.
- Hegele RA, Cao H, Anderson CM. et al. Heterogeneity of nuclear lamin A mutations in Dunnigan-type familial partial lipodystrophy. J Clin Endocrinol Metab. 2000;85:3431–3435. [PubMed: 10999845]
- 79.
- Garg A, Vinaitheerthan M, Weatherall PT. et al. Phenotypic heterogeneity in patients with familial partial lipodystrophy (Dunnigan variety) related to the site of missense mutations in lamin A/C gene. J Clin Endocrinol Metab. 2001;86:59–65. [PubMed: 11231979]
- 80.
- Behrens GM, Lloyd D, Schmidt HH. et al. Lessons from lipodystrophy: LMNA, encoding lamin A/C, in HIV therapy- associated lipodystrophy. AIDS. 2000;14:1854–1855. [PubMed: 10985325]
- 81.
- Hegele RA, Cao H, Harris SB. et al. Genetic variation in LMNA modulates plasma leptin and indices of obesity in aboriginal Canadians. Physiol Genomics. 2000;3:39–44. [PubMed: 11015599]
- 82.
- Hegele RA, Huff MW, Young TK. Common genomic variation in LMNA modulates indexes of obesity in Inuit. J Clin Endocrinol Metab. 2001;86:2747–2751. [PubMed: 11397881]
- 83.
- Wolford JK, Hanson RL, Bogardus C. et al. Analysis of the lamin A/C gene as a candidate for type II diabetes susceptibility in Pima Indians. Diabetologia. 2001;44:779–782. [PubMed: 11440372]
- 84.
- Vantyghem MC, Millaire A, Cuisset JM. et al. Muscular and cardiac phenotype in patients affected by the familial partial lipodystrophy (FPLD) or Dunnigan syndrome First International Workshop on Lipoatrophic diabetes and other syndromes of lipodystrophiesMarch 2001, Bethesda, USA, abstract P22.
- 85.
- Sullivan T, Escalante-Alcalde D, Bhatt H. et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–920. [PMC free article: PMC2169344] [PubMed: 10579712]
- 86.
- Raharjo WH, Enarson P, Sullivan T. Nuclear envelope defects associated with LMNA mutations causing dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J Cell Sci. 2001;114:4447–4457. [PubMed: 11792810]
- 87.
- Nagano A, Koga R, Ogawa M. et al. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat Genet. 1996;12:254–259. [PubMed: 8589715]
- 88.
- Ellis JA, Craxton M, Yates JR. et al. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J Cell Sci. 1998;111:781–792. [PubMed: 9472006]
- 89.
- Fairley EA, Kendrick-Jones J, Ellis JA. The Emery-Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. J Cell Sci. 1999; 112:2571–2582. [PubMed: 10393813]
- 90.
- Sabatelli P, Lattanzi G, Ognibene A. et al. Nuclear alterations in autosomal-dominant Emery-Dreifuss muscular dystrophy. Muscle Nerve. 2001;24:826–829. [PubMed: 11360268]
- 91.
- Fidzianska A, Toniolo D, Hausmanowa-Petrusewicz I. Ultrastructural abnormality of sarcolemmal nuclei in Emery-Dreifuss muscular dystrophy (EDMD). J Neurol Sci. 1998;159:88–93. [PubMed: 9700709]
- 92.
- Ognibene A, Sabatelli P, Petrini S. et al. Nuclear changes in a case of X-linked Emery-Dreifuss muscular dystrophy. Muscle Nerve. 1999;22:864–869. [PubMed: 10398203]
- 93.
- Vigouroux C, Auclair M, Dubosclard E. et al. Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in lamin A/C gene. J Cell Sci. 2001; 114:4459–4468. [PubMed: 11792811]
- 94.
- Holt I, Clements L, Manilal S. et al. The R482Q lamin A/C mutation that causes lipodystrophy does not prevent nuclear targeting of lamin A in adipocytes or its interaction with emerin. Eur J Hum Genet. 2001;9:204–208. [PubMed: 11313760]
- 95.
- Östlund C, Bonne G, Schwartz K. et al. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J Cell Sci. 2001;114:4435–4445. [PubMed: 11792809]
- 96.
- Mak YF, Ponder BA. RET oncogene. Curr Opin Genet Dev. 1996; 6:82–86. [PubMed: 8791480]
- 97.
- Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. 1996;271:14653–14656. [PubMed: 8663349]
- 98.
- Galy V, Olivo-Marin JC, Scherthan H. et al. Nuclear pore complexes in the organization of silent telomeric chromatin. Nature. 2000;403:108–112. [PubMed: 10638763]
- 99.
- Gasser SM. Positions of potential: nuclear organization and gene expression. Cell. 2001;104:639–642. [PubMed: 11257217]
- 100.
- Cohen M, Lee KK, Wilson KL. et al. Transcriptional repression, apoptosis, human disease and the functional evolution of the nuclear lamina. Trends Biochem Sci. 2001;26:41–47. [PubMed: 11165516]
- 101.
- Hutchison CJ, Alvarez-Reyes M, Vaughan OA. Lamins in disease: why do ubiquitously expressed nuclear envelope proteins give rise to tissue-specific disease phenotypes? J Cell Sci. 2001; 114:9–19. [PubMed: 11112685]
- 102.
- Wilson KL, Zastrow MS, Lee KK. Lamins and disease: insights into nuclear infrastructure. Cell. 2001;104:647–650. [PubMed: 11257219]
- 103.
- Buendia B, Santa-Maria A, Courvalin JC. Caspase-dependent proteolysis of integral and peripheral proteins of nuclear membranes and nuclear pore complex proteins during apoptosis. J Cell Sci. 1999; 112:1743–1753. [PubMed: 10318766]
- 104.
- Guillemin K, Williams T, Krasnow MA. A nuclear lamin is required for cytoplasmic organization and egg polarity in Drosophila. Nat Cell Biol. 2001;3:848–851. [PubMed: 11533666]
- 105.
- Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci USA. 1997;94:849–854. [PMC free article: PMC19602] [PubMed: 9023345]
- 106.
- De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, kassouri N. et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726–736. [PMC free article: PMC384949] [PubMed: 11799477]
- 107.
- Roberts R, Schwartz K. Myocardial diseases. Circulation. 2000;102:IV34–IV39. [PubMed: 11080129]
- Introduction
- Disorders of Cardiac and/or Skeletal Muscles Linked to LMNA Alterations
- Lipodystrophies and the Familial Partial Lipodystrophy of the Dunnigan Type (FPLD)
- Familial Partial Lipodystrophy of the Dunnigan Type (FPLD)
- Could Some Patients with LMNA Mutations be Affected by Both Skeletal or Cardiac Muscular Symptoms and Lipodystrophy?
- Experimental Models of Lamin A/C Alterations
- Nuclear Alterations in Cells Harboring LMNA Mutations
- Conclusion
- Addendum
- Acknowledgments
- References
- Laminopathies: One Gene, Two Proteins, Five Diseases.. - Madame Curie Bioscience...Laminopathies: One Gene, Two Proteins, Five Diseases.. - Madame Curie Bioscience Database
- Structural and Functional Relation of Neuropilins - Madame Curie Bioscience Data...Structural and Functional Relation of Neuropilins - Madame Curie Bioscience Database
- Fibroblast Growth Factors - Madame Curie Bioscience DatabaseFibroblast Growth Factors - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...