Summary
Clinical characteristics.
Isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD) is characterized by inappropriately low serum concentrations of the gonadotropins LH (luteinizing hormone) and FSH (follicle-stimulating hormone) in the presence of low circulating concentrations of sex steroids. IGD is associated with a normal sense of smell (normosmic IGD) in approximately 40% of affected individuals and an impaired sense of smell (Kallmann syndrome) in approximately 60%. IGD can first become apparent in infancy, adolescence, or adulthood. Infant boys with congenital IGD often have micropenis and cryptorchidism. Adolescents and adults with IGD have clinical evidence of hypogonadism and incomplete sexual maturation on physical examination. Adult males with IGD tend to have prepubertal testicular volume (i.e., <4 mL), absence of secondary sexual features (e.g., facial and axillary hair growth, deepening of the voice), decreased muscle mass, diminished libido, erectile dysfunction, and infertility. Adult females have little or no breast development and primary amenorrhea. Although skeletal maturation is delayed, the rate of linear growth is usually normal except for the absence of a distinct pubertal growth spurt.
Diagnosis/testing.
IGD is typically diagnosed in adolescents presenting with absent or partial puberty using biochemical testing that reveals low serum testosterone or estradiol (hypogonadism) that results from complete or partial absence of GnRH-mediated release of LH and FSH (hypogonadotropic hypogonadism [HH]) in the setting of otherwise normal anterior pituitary anatomy and function and in the absence of secondary causes of HH. Pathogenic variants in more than 25 genes account for about half of all IGD; the genetic cause for the remaining cases of IGD is unknown.
Management.
Treatment of manifestations: To induce and maintain secondary sex characteristics, gradually increasing doses of testosterone or human chorionic gonadotropin (hCG) injections in males or estrogen and progestin in females; to stimulate spermatogenesis or folliculogenesis, either combined gonadotropin therapy (hCG and human menopausal gonadotropins [hMG] or recombinant FSH) or pulsatile GnRH therapy. If conception fails despite spermatogenesis in a male or ovulation induction in a female, in vitro fertilization may be an option.
Prevention of secondary complications: Optimal calcium and vitamin D intake should be encouraged and specific treatment for decreased bone mass as needed.
Surveillance: For children of both sexes with findings suggestive of IGD, monitor at regular intervals after age 11 years: sexual maturation (by Tanner staging on physical examination); gonadotropin and sex hormone levels; bone age. In individuals with confirmed IGD, monitor at regular intervals: serum sex steroid levels (to guide optimal hormone replacement); bone mineral density.
Evaluation of relatives at risk: If the pathogenic variant(s) in a family are known, genetic testing of prepubertal at-risk relatives may be indicated to clarify their genetic status. Because of variable expressivity, a prepubertal child with a known pathogenic variant may progress through puberty in a normal or delayed fashion, or not at all; therefore, clinical reevaluation over time is necessary.
Genetic counseling.
IGD can be inherited in an X-linked, autosomal dominant, or autosomal recessive manner. Almost all IGD-related genes have also been associated with indeterminate or oligogenic inheritance. Recurrence risk counseling is based on family history and the results of molecular genetic testing when available. Carrier testing for at-risk relatives in families with X-linked IGD or autosomal recessive IGD is possible if the pathogenic variant(s) in the family are known. Prenatal testing for a pregnancy at increased risk is possible if the pathogenic variant(s) in the family are known.
GeneReview Scope
Table
Normosmic isolated gonadotropin-releasing hormone deficiency Kallmann syndrome
Diagnosis
Isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD) can be associated with a normal sense of smell (normosmic IGD) or an impaired sense of smell (Kallmann syndrome [KS]).
Suggestive Findings
Isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD) should be suspected in individuals with the following:
- Absent or partial puberty at presentation in adolescents; low serum testosterone or estradiol on biochemical testing
- Findings of incomplete sexual maturation on physical examination as determined by Tanner staging (see Table 1):
- Men with IGD typically have Tanner stage I-II genitalia (prepubertal testicular volumes; i.e., <4 mL); however, some males show evidence of partial pubertal maturation [Pitteloud et al 2001].
- Women with IGD typically have Tanner stage I breast development and amenorrhea; however, some have spontaneous breast development and occasional menses [Shaw et al 2011].
- Both men and women with IGD typically have Tanner stage II-III pubic hair, since pubic hair is controlled in part by adrenal androgens.
In rare males, IGD may present later in adulthood (i.e., adult-onset IGD). However, in these individuals, as puberty was not disrupted, sexual maturation is complete and secondary sexual characteristics may be fully developed. Diagnosis of adult-onset IGD relies on documentation of hypogonadotropic hypogonadism (HH) and absence of other secondary causes of HH. - Total testosterone (T) <100 ng/dL in males and estradiol (E2) <50 pg/mL in females
- Inappropriately low or normal serum concentration of LH (luteinizing hormone) and FSH (follicle stimulating hormone) in the presence of low circulating concentrations of sex steroids. Levels of other anterior pituitary hormones are typically normal.
- Imaging findings of IGD
- In persons with IGD: typically, normal-appearing hypothalamus and pituitary on MRI exam
- In persons with KS: typically, aplasia or hypoplasia of the olfactory bulbs/sulci/tracts.
- Olfactory findings. Olfactory function is evaluated by history and by formal diagnostic smell tests, such as the University of Pennsylvania smell identification test (UPSIT), a "scratch and sniff" test that evaluates an individual's ability to identify 40 microencapsulated odorants and can be easily performed in most clinical settings [Doty 2007]. Anosmia, hyposmia, or normosmia is identified using the UPSIT manual normogram, which incorporates an individual's score, age at testing, and sex.Individuals with IGD with either self-reported complete anosmia or a score of hyposmia/anosmia on UPSIT testing are diagnosed with KS, while those with normal olfactory function are diagnosed with normosmic IGD (nIGD) [Lewkowitz-Shpuntoff et al 2012].

Figure 1.
Testing algorithm to establish the diagnosis of isolated GnRH deficiency (IGD) in males

Figure 2.
Testing algorithm to establish the diagnosis of isolated GnRH deficiency (IGD) in females
Table 1.
Tanner Staging
Establishing the Diagnosis
The diagnosis of IGD is established in a proband based on clinical and biochemical investigations above; a genetic diagnosis can be made with identification of pathogenic (or likely pathogenic) variant(s) in one of the genes listed in Table 2a and Table 2b.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
See Table 2a for the most common genetic causes (i.e., pathogenic variants of any one of the genes included in this table account for >2% of IGD) and Table 2b for less common genetic causes (i.e., pathogenic variants of any one of the genes included in this table are reported in only a few families).
Molecular testing approaches can include serial single-gene testing, use of a multigene panel, and more comprehensive genomic testing.
Serial single-gene testing can be considered based on mode of inheritance and clinical findings, especially non-reproductive phenotypic features that indicate that pathogenic variation of a particular gene is most likely. Sequence analysis of the gene of interest is performed first, followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
To help prioritize the order of serial single-gene testing, the following can be considered (see Figure 3 and Figure 4):
Figure 3.
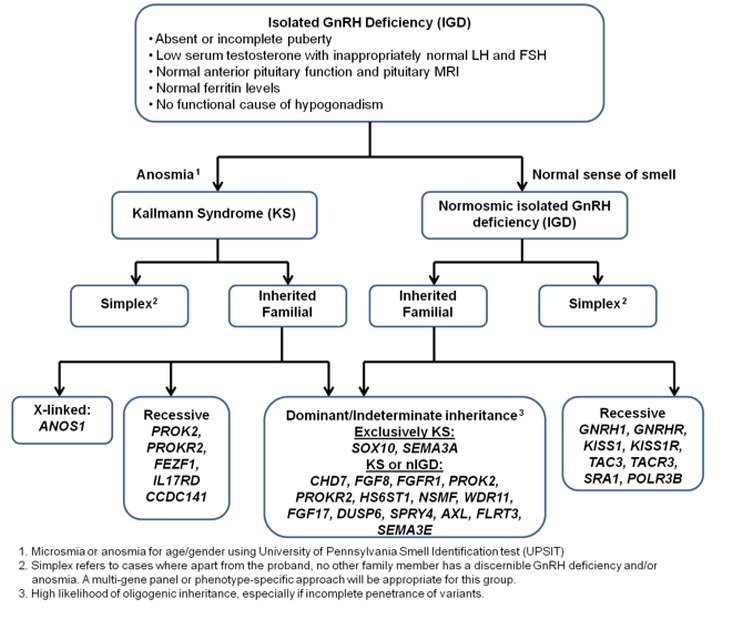
Genes associated with isolated GnRH deficiency (IGD) by sense of smell and mode of inheritance
Figure 4.
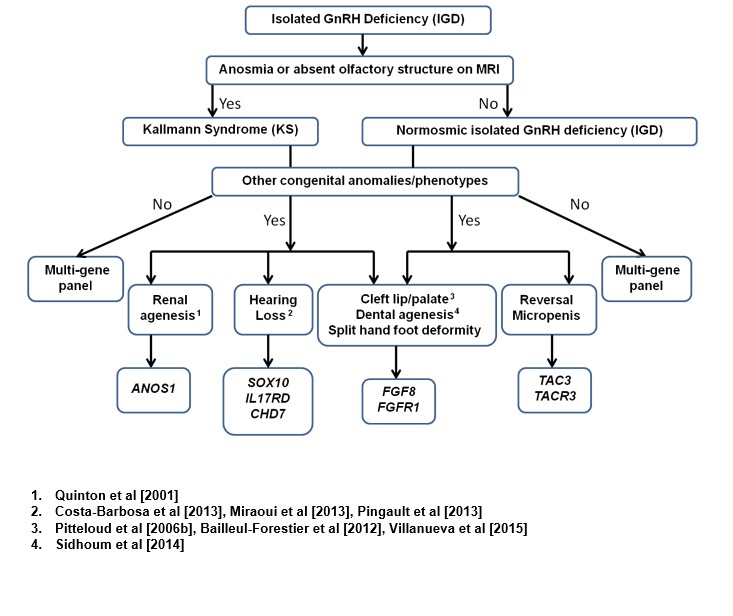
Suggested guidelines for prioritization of genetic testing for persons with IGD based on phenotype [modified from Au et al 2011]
- Sense of smell
- Pathogenic variants in CHD7, FGF8, FGF17, FGFR1, HS6ST1, NSMF (NELF), PROK2, PROKR2, and WDR11 cause both Kallmann syndrome (KS) and normosmic IGD (nIGD).
- Pathogenic variants in ANOS1 (KAL1), CCDC141, FEZF1, IL17RD, SEMA3A, SEMA3E, and SOX10 cause KS.
- Pathogenic variants in GNRH1, GNRHR, KISS1, KISS1R (GPR54), TAC3, and TACR3 cause nIGD.
- Mode of inheritance
- X-linked. Sequence analysis of ANOS1 (KAL1) is the highest-yield molecular genetic test.
- Autosomal dominant. In families with clear autosomal dominant inheritance, testing of CHD7, FGFR1, FGF8 and SOX10 can be considered.
- Autosomal recessive. Testing of GNRH1, GNRHR, KISS1, KISS1R, TAC3, and TACR3 can be considered in families with autosomal recessive normosmic IGD; testing of FEZF1, PROK2 and PROKR2 can be considered in families with autosomal recessive KS.
- Associated phenotypic features. The presence of some associated clinical phenotypic features may also help prioritize genetic testing in IGD [Costa-Barbosa et al 2013]. See Figure 4.
A multigene panel that includes the genes listed in Table 2a and Table 2b and other genes of interest (see Differential Diagnosis) may be considered. This should be the first step when the proband has no clearly affected family members and/or no associated phenotypic features. Note: The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2a.
Molecular Genetic Testing Used in Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency: Most Common Genetic Causes
Table 2b.
Molecular Genetics of Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency: Less Common Genetic Causes
Clinical Characteristics
Clinical Description
The clinical manifestations of isolated GnRH deficiency (IGD) depend on the stage of development at which the deficiency in the reproductive axis first occurred – in infancy, adolescence, or (rarely) adulthood. Most individuals with IGD are identified at puberty; however, suggestive clinical features may be present in infancy.
Reproductive Phenotype
Infancy. Microphallus (stretched penile length <1.9 cm in a full-term newborn male) and cryptorchidism (undescended testes) represent two early clinical findings that may be present in male infants with IGD, although the significance of these findings is usually not recognized until puberty. Both clinical features reflect congenital GnRH deficiency and, if measured in male infants, concentrations of testosterone, LH (luteinizing hormone), and FSH (follicle stimulating hormone) are low in the first six months of life in these infants (neonatal window) [Grumbach 2005].
Although microphallus and cryptorchidism can occur in both forms of IGD (KS and normosmic IGD), these features are more common in males with KS than in those with normosmic IGD [Pitteloud et al 2002a].
Female infants typically do not exhibit any clinical features that may indicate IGD.
Adolescence. At puberty, most individuals with IGD have abnormal sexual maturation, usually with incomplete development of secondary sexual characteristics. However, the degree to which sexual maturation is affected can vary (see Fertile hypogonadal variant of IGD in males).
Males with IGD typically have prepubertal testicular volume (i.e., <4 mL), absence of secondary sexual features (e.g., facial and axillary hair growth and deepening of the voice), and decreased muscle mass.
Females with IGD typically have little or no breast development and primary amenorrhea, but milder presentations with spontaneous menses are recognized [Shaw et al 2011].
Since adrenal maturation proceeds normally, the low levels of androgens produced in the adrenal glands may be sufficient for normal onset of pubic hair growth (adrenarche) in both sexes.
Because of the failure of growth plates in the bone to fuse in the absence of sex hormones, most individuals with IGD, both males and females may have disproportionate arm span compared to height (arm span typically exceeds height by ≥5 cm). Whereas skeletal maturation is delayed, the rate of linear growth is usually normal (save for the absence of a distinct pubertal growth spurt) [Van Dop et al 1987].
Fertile hypogonadal variant of IGD in males. Some degree of pubertal development can occur in some individuals with IGD. The relatively mildest form of abnormal pubertal development is found in males who have clinical evidence of hypogonadism with low serum concentration of testosterone but evidence of partial pubertal development with normal or near-normal testicular volumes, normal levels of inhibin B (the seminiferous tubular secretory protein), and, often, sperm in their ejaculate.
Reversal of IGD, defined as restoration of normal serum testosterone concentrations after cessation of even brief treatment with sex steroid, gonadotropin, or GnRH, occurs in about 10% of all men with IGD, including those with KS [Raivio et al 2007, Sidhoum et al 2014]. This post-treatment “awakening” of the hypothalamo-pituitary-gonadal (HPG) axis suggests the presence of hypothalamic GnRH neurons that do not function during adolescence and possibly require hitherto undefined stimuli (potentially environmental/sex steroid exposure) to initiate normal activity. The precise physiologic basis of the reversal phenomenon is yet to be fully understood.
Olfactory Phenotype
Anosmia. The impaired olfactory function in Kallmann syndrome can be either hyposmia or complete anosmia) [Bianco & Kaiser 2009]. The difference between hyposmia and anosmia is quantitative and not qualitative (i.e., odorants can be variably affected in persons with hyposmia). Most individuals with impaired smell do not have any physical or social impairment and the finding often goes unnoticed until IGD is diagnosed.
Pathophysiology
Pulsatile secretion of GnRH into the hypophyseal portal circulation represents the initial neuroendocrine step in the regulation of the hypothalamo-pituitary-gonadal (HPG) axis in both sexes. Thus, this specialized GnRH neuronal network plays a commanding role in this biologic hierarchy and controls episodic gonadotropin secretion, modulates gonadal steroid feedback, and ultimately determines the initiation or suppression of pubertal development and fertility across the life cycle [Hoffman & Crowley 1982, Crowley et al 1985].
Under normal conditions, the GnRH neuronal network undergoes a series of dynamic changes from fetal life to adulthood. The initiation of GnRH secretion is initiated in early fetal life and remains active until the first several months of infancy (representing the "mini-puberty"), and then becomes remarkably dampened during the years of the childhood "quiescence" [Waldhauser et al 1981]. At puberty, unknown biologic triggers re-ignite GnRH secretion, resulting in full sexual maturation. Therefore, the controls of the reproductive axis are in dynamic flux, turning on and turning off in response to as-yet-unknown biologic signals at various points in the reproductive life cycle.
In individuals with IGD, analyses of the pulsatile pattern of gonadotropins have demonstrated a rather broad spectrum of abnormal developmental patterns varying from completely absent GnRH-induced LH pulses to sleep-entrained GnRH release that is indistinguishable from that of early puberty [Spratt et al 1987, Nachtigall et al 1997, Raivio et al 2007]. This broad spectrum of neuroendocrine activity accounts for the variable reproductive phenotypes observed in persons with IGD.
Genotype-Phenotype Correlations
Gene-specific phenotypes have been noted; see Table 3 and Figure 4.
No reproductive or non-reproductive phenotype is specific to a single pathogenic variant or particular type of pathogenic variant in any of the IGD-related genes.
Penetrance
The underlying genetic etiology typically determines the penetrance of both reproductive and non-reproductive phenotypes.
The penetrance for the KS phenotype (both IGD and anosmia) is generally complete in males with an ANOS1 (KAL1) pathogenic variant. However, other non-reproductive phenotypes may have variable penetrance even in the setting of the same ANOS1 (KAL1) defect. One set of identical twin males with a small deletion in ANOS1 (KAL1) with discordant neuroendocrine and non-reproductive phenotypes has been documented: one twin had a ventricular septal defect and a much greater LH and FSH response to a serial LH-RH stimulation test, whereas the other had exotropia and a lower response to the serial LH-RH stimulation test [Matsuo et al 2000].
Penetrance for IGD is also fairly high when pathogenic variants occur in the biallelic state (i.e., recessive variants) (FEZF1, GNRH1, GNRHR, IL17RD, KISS1, KISS1R, TAC3, TACR3). However, penetrance for the reproductive phenotype in those with pathogenic heterozygous variants in almost all known IGD-related genes is reduced, as individuals with normal gonadal function who are heterozygous for pathogenic variants in these genes have been documented. Discordance for KS has also been demonstrated in identical twins, suggesting that additional modifiers may play a role in phenotypic expression [Hipkin et al 1990].
Penetrance for anosmia in men with an ANOS1 (KAL1) pathogenic variant is generally complete, whereas penetrance for abnormal olfactory function in individuals with IGD and heterozygous pathogenic variants in several genes (CHD7, FGFR1, FGF8, FGF17, HS6ST1, NSMF, PROK2, PROKR2) is reduced, as normosmia, hyposmia, or anosmia can be seen [Pitteloud et al 2006a, Cole et al 2008, Falardeau et al 2008, Miraoui et al 2013, Balasubramanian et al 2014].
Nomenclature
The biochemical term "hypogonadotropic hypogonadism" has evolved with the increased understanding of reproductive physiology.
The term “hypogonadism” refers to impaired sexual development based on findings from the individual's clinical history (e.g., amenorrhea, hot flashes, erectile dysfunction) as well as physical examination (e.g., small testes, vaginal pallor).
With greater understanding of the hypothalamo-pituitary-gonadal (HPG) axis (see Pathophysiology) and the introduction of urinary gonadotropin measurements, the term "hypergonadotropic" hypogonadism was used to identify those with a primary gonadal defect, while "hypogonadotropic" hypogonadism identified those with a central (i.e., pituitary or hypothalamic) defect.
When anatomic (and later functional) causes of central hypogonadism were identified, "idiopathic" or "isolated" hypogonadotropic hypogonadism (IHH) came into use to indicate those individuals in whom secondary causes of hypogonadotropic hypogonadism had been excluded.
Subsequently the ability to measure the effect of exogenous GnRH administration demonstrated that the vast majority of individuals with "idiopathic" HH had a functional deficiency of GnRH resulting from a defect in GnRH biosynthesis, secretion, and/or action (hence "isolated GnRH deficiency" [IGD]). Aside from hypothalamic GnRH deficiency, individuals with IGD typically have normal pituitary function tests and their hypogonadism typically responds to a physiologic regimen of exogenous GnRH [Hoffman & Crowley 1982].
At this point, the term "isolated GnRH deficiency" (IGD) more properly reflects the current understanding of the clinical entity rather than the previous biochemical description of IHH and, thus, is the better term for what was previously called IHH.
Prevalence
A recent epidemiologic study in Finland showed a minimal incidence of KS of 1:30,000 in males and 1:125,000 in females [Laitinen et al 2011].
In the authors' cohort of 250 individuals with IGD, males predominate with a male-to-female ratio of nearly 4:1 [Seminara et al 1998].
KS accounts for nearly two thirds of individuals with isolated GnRH deficiency (IGD).
Genetically Related (Allelic) Disorders
CHD7. Heterozygous pathogenic variants or microdeletions in CHD7 can also cause CHARGE syndrome. The syndrome is characterized by (and derives its name from) coloboma, heart abnormalities, choanal atresia, retardation of growth and development, genital hypoplasia, and ear abnormalities [Vissers et al 2004]. The genital abnormalities in CHARGE syndrome are caused by hypogonadotropic hypogonadism and are frequently accompanied by olfactory defects and cleft lip/palate [Pinto et al 2005].
FGFR1. In addition to KS with highly variable expressivity, other phenotypes associated with germline pathogenic variants in FGFR1 include Pfeiffer syndrome type 1 and osteoglophonic dwarfism.
- Of the pathogenic variants that cause Pfeiffer syndrome, 95% occur in FGFR2 and only 5% occur in FGFR1 (see FGFR-Related Craniosynostosis Syndromes). Pfeiffer syndrome type 1 is characterized by coronal craniosynostosis with moderate-to-severe midface hypoplasia, usually normal intellect, broad and medially deviated thumbs, and great toes with variable degree of brachydactyly. Hearing loss and hydrocephalus can be seen on occasion.
- Osteoglophonic dwarfism involves rhizomelic dysplasia, dysmorphic facial features, fibrous dysplasia, and clover-leaf skull.
Mosaic activating pathogenic variants in FGFR1 are associated with encephalocraniocutaneous lipomatosis (ECCL). The pathogenic variants reported in ECCL are of postzygotic origin but arise early during development. ECCL comprises a spectrum of predominantly congenital anomalies. In its typical form, ECCL is characterized by congenital skin, eye, and brain anomalies, in particular intracranial and spinal lipomas.
SOX10. Heterozygous pathogenic variants or microdeletions in SOX10 can also cause Waardenburg syndrome, without IGD. The syndrome is characterized by deafness, skin/hair/iris hypopigmentation, Hirschsprung disease, and neurologic defects [Pingault et al 2013].
Differential Diagnosis
Other Causes of Hypogonadotropic Hypogonadism
Hypogonadotropic hypogonadism refers to a diverse group of clinical conditions with characteristic biochemical findings of inappropriately low serum concentrations of LH (luteinizing hormone) and FSH (follicle stimulating hormone) occurring in the setting of hypogonadism.
Distinguishing between isolated GnRH deficiency (IGD) and secondary causes of hypogonadotropic hypogonadism and syndromic/genetic causes of hypogonadotropic hypogonadism often requires additional clinical, laboratory, and radiologic evaluations. These may include physical examination for other systemic findings, family history, and measurement of serum concentration of other pituitary hormones, serum iron studies, and hypothalamic/pituitary imaging. Of note, despite a thorough evaluation, IGD can sometimes be difficult to distinguish from other causes of decreased gonadotropin secretion. Hence, molecular genetic testing of the known IGD-related genes (Table 2) may help make the diagnosis of IGD
Acquired causes. Multiple disease processes ranging from systemic diseases to brain and pituitary tumors can result in impaired gonadotropin secretion. These conditions, which can be relatively common and frequently give rise to defects in other pituitary hormones, include the following:
- CNS or pituitary tumors
- Pituitary apoplexy
- Brain/pituitary radiation
- Head trauma
- Drugs: GnRH agonists/antagonists, glucocorticoids, narcotics, chemotherapy, drugs causing hyperprolactinemia
- Functional deficiency resulting from hyperprolactinemia, chronic systemic illness, eating disorders, malnutrition, hypothyroidism, diabetes mellitus, Cushing's disease
- Systemic diseases such as sarcoidosis and histiocytosis
Syndromes listed in Table 4 can be associated with hypogonadotropic hypogonadism along with other significant clinical findings and/or other pituitary hormone deficits.
Table 4.
Syndromes Associated with Hypogonadotropic Hypogonadism
Differential Diagnosis of IGD at Specific Developmental Stages
Infancy. Although males with IGD may have cryptorchidism and/or microphallus at birth, these features are not specific for IGD. Numerous disorders can give rise to these genital defects, ranging from isolated findings to syndromes such as Prader-Willi syndrome or abnormal pituitary development (see PROP1-Related Combined Pituitary Hormone Deficiency). This is particularly true for cryptorchidism, the most common birth defect of the male genitalia.
Adolescence. Perhaps the most difficult distinction to make is between IGD and constitutional delay of puberty (CDP). Time is a critical factor in distinguishing between these two conditions. In CDP, spontaneous and otherwise normal puberty eventually occurs, whereas in IGD spontaneous sexual maturation does not occur at any time. Evidence suggests that CDP and IGD are not discrete clinical entities but rather are part of a phenotypic spectrum. In families with IGD, delayed puberty occurs at a much higher frequency in otherwise "normal" family members than in the general population, suggesting that CDP may represent a milder clinical variant of the IGD phenotype [Waldstreicher et al 1996, Pitteloud et al 2006a].
Although the distinction between CDP and IGD cannot be reliably made at any age, age 18 years has traditionally been suggested as the age at which IGD can be diagnosed; however, the recent description of IGD "reversal" occurring in persons in their 20s and later raises the possibility that such individuals may have a severe form of CDP [Raivio et al 2007]. In contrast, the presence of other clinical features associated with IGD (e.g., anosmia, synkinesia) (Table 3) may result in a diagnosis of IGD being made before age 18 years.
Currently, no clinically available test can reliably differentiate CDP from IGD. Data analyses have suggested that the mean serum concentrations of LH and sex hormones after GnRH or hCG (human chorionic gonadotropin) stimulation vary significantly between individuals with CDP and those with IGD. However, the clinical utility of measuring serum LH and sex hormone concentrations after stimulation with GnRH and hCG is limited by the significant variation in individual LH and sex hormone serum concentrations, resulting in considerable overlap between groups [Degros et al 2003].
A peak-to-basal ratio of free alpha subunit (FAS) after the administration of GnRH has been proposed to help distinguish between CDP and IGD with a sensitivity and specificity in the 95% range and an overlap rate of 10% [Mainieri & Elnecave 2003]. More recently, combining a 19-day hCG test with a conventional GnRH test has also been proposed to improve the differentiation between IGD and CDP [Segal et al 2009]. However, given the relatively small number of individuals studied and limited follow-up in both studies, prospective validation is required to determine the true diagnostic reliability of the above tests.
Figure 1 and Figure 2 provide testing algorithms for establishing the diagnosis of isolated GnRH deficiency in males and females, respectively.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with IGD, the following evaluations are recommended:
- Assessment of laboratory findings* of hypogonadotropic hypogonadism if not already performed as part of the diagnostic work up* Serum concentration of LH (luteinizing hormone) and FSH (follicle-stimulating hormone) and in males total testosterone (T) <100 ng/dL and in females estradiol (E2) <50 pg/mL
- Assessment for presence of possible non-reproductive features including: renal ultrasound examination (to detect unilateral renal agenesis), hearing tests (to detect sensorineural hearing loss), skeletal survey (to detect limb/spine bony abnormalities), dental exam (to detect dental agenesis), eye exam (to detect iris and/or chorioretinal coloboma) and developmental assessment (if there is evidence of developmental delay)
- In addition to assessing the degree of hypogonadism/GnRH deficiency, potential deterioration in bone health that may have resulted from periods of low-circulating sex hormones needs to be addressed. Depending on the timing of puberty, duration of GnRH deficiency, and other osteoporotic risk factors (e.g., glucocorticoid excess, smoking), one should consider obtaining a bone mineral density study (see Prevention of Secondary Complications).
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
An expert European consensus statement on the management of IGD has recently been published [Boehm et al 2015] (full text).
Typically, a definitive diagnosis of IGD is made around age 18 years. Occasionally, however, a high clinical suspicion of IGD may be present in an adolescent presenting with anosmia and delayed puberty or in an infant with microphallus and cryptorchidism.
Males with IGD Age ≥18 Years
Treatment options include sex steroids, gonadotropins, and pulsatile GnRH administration. Choice of therapy in adults is determined by the goal(s) of treatment (i.e., to induce and maintain secondary sex characteristics and/or to induce and maintain fertility). The selection of hormone replacement therapy is also based on the preference of the individual being treated; however, when fertility is not immediately desired, replacement with testosterone therapy is the most practical option. As the majority of individuals with IGD have not progressed through puberty at the time of diagnosis, initial therapy should be started at low doses and gradually increased to adult doses once the development of secondary sexual characteristics is achieved.
Hormone replacement therapy for males not desiring fertility
- Testosterone therapy in the form of injectable and transdermal routes of testosterone administration is typically used to both induce puberty and maintain adult levels of testosterone. Recently nasal testosterone has become available but use has not been reported in individuals with IGD.The injectable testosterone preparations have a "roller-coaster" pharmacokinetic effect, with peak and trough levels that can go to extraphysiologic levels; thus, the transdermal preparations have the added benefit of offering a more favorable pharmacokinetic profile. A typical adult dose of testosterone replacement is 200 mg of testosterone ester injection every two weeks or 5 g of a 1% testosterone gel every day. Doses do vary with newer testosterone preparations; manufacturer's instructions should be followed for individual testosterone preparations.Men using topical androgen replacement must take care to avoid exposing other individuals to treated skin. Anecdotal reports suggest that the transmission of clinically effective levels of testosterone from the affected individual to other family members (including women and children) is possible, with undesirable side effects.Once puberty is initiated, testosterone replacement therapy is usually required indefinitely to ensure normal sexual function and maintenance of proper muscle, bone, and red blood cell mass.However, in approximately 10% of males, reversal of IGD may occur; thus, if clinical evidence shows endogenous activity of the hypothalamo-pituitary-axis (e.g., testicular growth on testosterone, maintained testosterone levels despite missing/withholding therapy), a brief washout of testosterone therapy should be done with monitoring of testosterone levels. If testosterone levels fall, therapy should be reinitiated. If levels are normal, no further testosterone therapy will be required; serial monitoring of levels should be undertaken, as some individuals may require reinitiation of therapy.
- Human chorionic gonadotropin (hCG). An alternative to testosterone therapy, hCG injections promote testicular growth, normalize serum concentration of testosterone, and induce development of secondary sexual characteristics.In adults, treatment with hCG is usually initiated at 1,500 IU intramuscularly or subcutaneously every other day to normalize serum testosterone concentrations. Dose should be increased by increments of 250 IU if serum testosterone levels remain low.Treatment with hCG must be weighed against the increased risk of developing gynecomastia (resulting from the estrogen produced by stimulation of the testes with hCG). To some extent the risk of gynecomastia can be minimized by gradually reducing the dose of hCG to the minimum required to sustain a serum testosterone concentration in the mid-normal range (~500 ng/dL).
Male Infants/Adolescents with Suspicion of IGD
If IGD is clinically suspected (e.g., low testosterone levels with low/normal gonadotropins) low-dose testosterone or hCG therapy can be given in early infancy to boys with microphallus to increase penile length [Bin-Abbas et al 1999, Young 2012].
Since a definitive diagnosis of IGD may not be possible until age 18 years, after infancy these boys do not generally need to be treated until around the time of puberty. At this time, if a high suspicion of IGD remains (e.g., associated anosmia and delay in onset puberty), these subjects may benefit from early initiation of hormonal replacement therapy with either testosterone or hCG treatment early in the pubertal period. A suggestive puberty induction regimen in adolescents is to start a long-acting testosterone ester at a dose of 25-50 mg, given intramuscularly every two weeks. The doses can be gradually increased by 25-50 mg every two to three months until full virilization is achieved. Once adult doses (~200 mg/2 weeks) are reached, further adjustments are based on serum testosterone levels.
Hormone replacement therapy for males desiring fertility (fertility induction in males). As testosterone replacement therapy suppresses spermatogenesis in the testes, gonadotropins or pulsatile GnRH therapy is usually required to realize the fertility potential in males.
- Gonadotropin therapy. In most males with IGD, a combination of gonadotropins (hCG along with either human menopausal gonadotropins [hMG] or recombinant FSH) is used to stimulate spermatogenesis. In males with very low testicular volumes (≤~8 mL) the initiating dose of hCG is usually 1,500 IU intramuscularly or subcutaneously every other day; FSH is added at doses ranging from 37.5 to 75 IU as either hMG or recombinant formulation. Trough serum testosterone concentrations (target: mid-normal range [~500 ng/dL]), trough serum FSH levels (target: mid-normal reference range), and sperm count are monitored to assess response. Recent trials show that in those with lower testicular volumes, priming with FSH prior to combination therapy may improve spermatogenic outcomes [Dwyer et al 2013].In males with higher baseline testicular volumes, treatment with hCG alone may be sufficient to achieve spermatogenesis and conception [Burris et al 1988]. However, if after six to nine months, semen analysis reveals persistent azoospermia or marked oligospermia, FSH is added to the regimen at doses ranging from 37.5 to 75 IU as either hMG or a recombinant formulation.In either treatment, testicular volume must be tracked, as this is one of the primary determinants of successful spermatogenesis. In fact, sperm are rarely seen in the semen analysis until testicular volume reaches 8 mL [Whitcomb & Crowley 1990]. In most males without a history of cryptorchidism, sperm function is usually normal and conception can occur even with relatively low sperm counts. Note: Liu et al [2009] have noted that previous treatment with gonadotropins may reduce the period of subsequent gonadotropin treatment required for initiation of spermatogenesis.If a pituitary defect exists, gonadotropin therapy becomes the treatment of choice.
- Pulsatile GnRH stimulation vs gonadotropin therapy. While either gonadotropin therapy or pulsatile GnRH stimulation can induce spermatogenesis in approximately 90%-95% of men with IGD, some men have a better response to pulsatile GnRH stimulation than to gonadotropin therapy.Subcutaneous administration of GnRH in a pulsatile manner through a portable pump that delivers a GnRH bolus every two hours is an efficient way of inducing testicular growth and spermatogenesis [Pitteloud et al 2002b]. As the primary defect of IGD is typically localized to the hypothalamus, the pituitary responds appropriately to physiologic doses of GnRH. Note: In the US, pulsatile GnRH therapy is not currently approved by the Food and Drug Administration for the treatment of infertility in men and, thus, is available for such treatment only at specialized research centers.
Females with IGD
Hormone replacement therapy for females not desiring fertility. Although a definitive diagnosis of IGD in females is usually made around age 18 years, occasionally a high clinical suspicion of IGD may be present in an adolescent presenting with anosmia and delayed puberty, and therapy may need to be initiated earlier (age ~14 years)
- To allow optimal breast development, initial treatment should consist of unopposed estrogen replacement via oral or topical preparations. Many formulations of estrogens are available; a suggested oral regimen is using premarin 0.3 mg daily to be increased gradually to an adult replacement dose of 1-1.25 mg daily.
- Once breast development is optimal, a progestin should be added for endometrial protection (e.g., cyclical Prometrium® 200 mg daily for 10-12 days).
- Although preference of the individual plays an important role in choice of treatment plan, low-estrogen formulations should be considered in women with clotting abnormalities (see Factor V Leiden Thrombophilia and Prothrombin Thrombophilia).
Hormone replacement therapy for females desiring fertility (fertility induction in females). Pulsatile GnRH stimulation and exogenous gonadotropins are FDA approved for folliculogenesis in women with IGD. Either therapy should be administered with close supervision by physicians specializing in ovulation induction. Intravenous administration of GnRH at various frequencies throughout the menstrual cycle closely mimics the dynamics of normal menstrual cycles resulting in ovulation of a single follicle [Santoro et al 1986]. This therapy offers a clear advantage over the traditional treatment with exogenous gonadotropins, which results in higher rates of both multiple gestation and ovarian hyperstimulation syndrome. For either approach, however, the rate of conception is approximately 30% per ovulatory cycle [Martin et al 1990].
Fertility Options in Individuals with IGD If Fertility Induction Is Unsuccessful
In vitro fertilization. Although successful spermatogenesis can be obtained in most males with IGD through pulsatile GnRH therapy or combined gonadotropin therapy, some men with KS caused by an ANOS1 (KAL1) pathogenic variant may have an atypical response to therapy [Sykiotis et al 2010a]. In those who respond to therapy, low sperm numbers can often result in conception; however, if infertility continues despite successful spermatogenesis or if spermatogenesis fails to occur, in vitro fertilization (IVF) is an option.
Similarly, if spontaneous conception fails to occur in women with IGD who have undergone ovulation induction, IVF may be an option.
Prevention of Secondary Complications
Optimal calcium and vitamin D intake should be encouraged and specific treatment for decreased bone mass with bisphosphonates should be considered depending on the degree of bone mineralization (see Evaluations Following Initial Diagnosis).
Surveillance
Children of both sexes with findings suggestive of IGD (e.g., microphallus, anosmia) should be monitored at regular intervals from age 11 years onwards with the following:
- Assessment of sexual maturation by Tanner staging (Table 1)
- Measurement of serum concentrations of LH, FSH, and total testosterone (T) in males and estradiol (E2) in females
- Bone age determinations
In individuals with a confirmed diagnosis of IGD, serum sex steroid levels (to guide optimal hormone replacement) and bone mineral density should be monitored at regular intervals.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from surveillance and prompt initiation of treatment. Evaluations can include:
- Molecular genetic testing if the pathogenic variant(s) in the family are known. However, a prepubertal child with a known pathogenic variant may progress through puberty in a normal fashion, delayed fashion, or not at all. Therefore, reevaluation of such individuals over time is important, and hormone treatment should be initiated only when IGD with impaired pubertal development is diagnosed.
- If the pathogenic variant(s) in the family are not known, clinical review of at-risk relatives of pubertal age to assess clinical onset of signs of puberty and if delayed, to initiate appropriate therapy for pubertal induction.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD) can be inherited in an X-linked, autosomal dominant, or autosomal recessive manner (see Figure 3).
Almost all IGD-related genes have also been associated with indeterminate or oligogenic inheritance (especially when an IGD-related pathogenic variant occurs in the heterozygous state [Sykiotis et al 2010b, Hanchate et al 2012, Miraoui et al 2013]).
A three-generation family history should be obtained to understand the mode of inheritance of IGD to aid genetic testing and genetic counseling. Detailed histories including questions regarding consanguinity, reproductive features (e.g., microphallus and cryptorchidism, pubertal development, fertility/infertility), olfactory function (normal sense of smell, hyposmia, anosmia), and non-reproductive features (e.g., craniofacial abnormalities including cleft lip/palate/missing teeth, hearing loss, synkinesia of the digits, and renal agenesis) should be obtained for all family members. If other individuals with IGD or these associated findings are identified in the family, the mode of inheritance may become apparent. In the majority of individuals, however, no such family history is present.
X-Linked Inheritance – Risk to Family Members
Parents of a male proband
- The father of an affected male will not have the disease nor will he be hemizygous for the ANOS1 (KAL1) pathogenic variant; therefore, he does not require further evaluation/testing.
- In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote (carrier). Note: If a woman has more than one affected son and no other affected relatives and if the pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism (maternal germline mosaicism has not been reported to date for ANOS1).
- If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote (carrier) or the affected male may have a de novo ANOS1 pathogenic variant, in which case the mother is not a carrier. About 70% of affected males represent simplex cases.
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
- If the mother of the proband has a ANOS1 pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Male sibs who inherit the pathogenic variant will be affected; female sibs who inherit the pathogenic variant will be heterozygous and may display some clinical features.
- If the proband represents a simplex case (i.e., a single occurrence in a family) and if the pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is slightly greater than that of the general population (though still <1%) because of the theoretic possibility of maternal germline mosaicism.
Offspring of a male proband
- With treatment, males with IGD can be fertile.
- Affected males transmit the ANOS1 pathogenic variant to:
- All of their daughters, who will be heterozygous and may display some clinical features.
- None of their sons.
Other family members. The proband's maternal uncles may be at risk of being affected and the maternal aunts may be at risk of being carriers for the ANOS1 pathogenic variant. The aunts' offspring, depending on their sex, may be at risk of being carriers or of being affected.
Heterozygote detection. Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the ANOS1 pathogenic variant has been identified in the proband.
Note: (1) Females heterozygous for an ANOS1 pathogenic variant may occasionally display clinical features that are diagnostic of IGD [Shaw et al 2011]. (2) Identification of female heterozygotes requires either (a) prior identification of the ANOS1 pathogenic variant in the family or, (b) if an affected male is not available for testing, molecular genetic testing first by sequence analysis, and if no pathogenic variant is identified, by gene-targeted deletion/duplication analysis.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
- Some individuals with autosomal dominant IGD have an affected parent.
- A proband with autosomal dominant IGD may have the disorder as the result of a de novo pathogenic variant. The proportion of cases caused by a de novo pathogenic variant is unknown.
- Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant include:
- A detailed pubertal history of both parents; and
- Molecular genetic testing of both parents for the pathogenic variant identified in the proband.
- If the pathogenic variant found in the proband cannot be detected in leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent (germline mosaicism for an IGD-related pathogenic variant has been reported in one family [Sato & Ogata 2006]).
- Evaluation of parents may determine that a parent has the pathogenic variant identified in the proband but has escaped previous diagnosis because of a milder phenotypic presentation, reduced penetrance, or late onset of the disorder. Therefore, an apparently negative family history cannot be fully confirmed until appropriate evaluations have been performed.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected and/or has a pathogenic variant, the risk to the sibs of inheriting the pathogenic variant is 50% although their actual risk may be lower due to variable expressivity and reduced penetrance.
- If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism [Sato & Ogata 2006].
Offspring of a proband. Each child of an individual with autosomal dominant IGD has a 50% chance of inheriting the pathogenic variant; however, the actual risk may be less than 50% because of variable expressivity and reduced penetrance.
Other family member. The risk to other family members depends on the genetic status of the proband's parents: if a parent is affected or has a known pathogenic variant, members of the parent's family may be at risk.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
- The parents of a child with autosomal recessive IGD are obligate heterozygotes (i.e., carriers of one pathogenic variant).
- Heterozygotes are typically unaffected.
- Occasionally, heterozygotes display clinical features diagnostic of IGD [Cole et al 2008, Gianetti et al 2010, Sykiotis et al 2010b, Chan et al 2011, Miraoui et al 2013]; this may be due to unknown environmental, epigenetic, or oligogenic factors.
Sibs of a proband
- At conception, each sib of an individual with autosomal recessive IGD has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Occasionally, heterozygotes display clinical features diagnostic of IGD [Cole et al 2008, Gianetti et al 2010, Sykiotis et al 2010b, Chan et al 2011, Miraoui et al 2013]; this may be due to unknown environmental, epigenetic, or oligogenic factors.
Offspring of a proband. The offspring of an affected individual are obligate heterozygotes (carriers) for an IGD-related pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an IGD-related pathogenic variant.
Carrier detection. Identification of heterozygotes requires prior identification of the IGD-related pathogenic variants in the family.
Indeterminate or Oligogenic Inheritance – Risk to Family Members
The exact risk for IGD to the family members of a simplex proband (a single individual with a disorder in a family) with IGD of unknown cause or a proband with an indeterminate or oligogenic inheritance is uncertain but may be up to 50%.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the IGD-related pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing for IGD are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Pituitary Network AssociationPO Box 1958Thousand Oaks CA 91358Phone: 805-499-9973Fax: 805-480-0633Email: info@pituitary.org
- The Pituitary FoundationUnited KingdomPhone: 0117 370 1320Email: enquiries@pituitary.org.uk
- The Pituitary SocietyVA Medical Center423 East 23rd StreetRoom 16048aWNew York NY 10010Phone: 212-951-7035Fax: 212-951-7050
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency: Genes and Databases
Table B.
OMIM Entries for Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency (View All in OMIM)
Molecular Pathogenesis
Over the last three decades, a combination of clinical investigational strategies and contemporary genetic approaches has revealed more than 25 causal/contributory genes for the nonsyndromic forms of IGD, with varied modes of inheritance. Broadly, two groups of genetic pathways are linked to IGD: (i) Neurodevelopmental genes govern the origin of the GnRH neurons and typically cause the Kallmann syndrome (KS) form of IGD. (ii) A group of neuroendocrine genes that control the secretion or action of GnRH cause the normosmic isolated GnRH deficiency (nIGD) form IGD. A small subset of genes may cause both KS and nIGD forms of IGD and this suggests that they govern both GnRH migration and GnRH secretion/action. (see Table 2a and Table 2b).
In addition to Mendelian modes of inheritance, a complex genetic architecture for IGD (occurring in 10%-15% of cases) has now been documented wherein pathogenic variants in two or more IGD-related genes are present in a single individual. These pathogenic variants are typically heterozygous and by themselves are not sufficient to cause IGD but require the presence of additional pathogenic variants in a second gene to cause IGD [Sykiotis et al 2010b]. Almost all of the known IGD-related genes have been associated with oligogenic inheritance. These oligogenic pathogenic variants presumably act in a synergistic manner, potentially accounting for some of the variable expressivity and reduced penetrance that is characteristic of IGD.
ANOS1 (KAL1)
Gene structure. ANOS1 (KAL1) comprises 14 exons and has no alternative spice variants.
Pathogenic variants. Reported pathogenic variants in ANOS1 include deletion of the entire gene, deletion of one or more exons, deletion of several nucleotides, pathogenic missense variants, pathogenic nonsense variants, and splice variants. For more information, see Table A.
Normal gene product. The protein encoded by ANOS1, anosmin 1, has 680 amino acids with functional similarities to molecules involved in neural development [Rugarli et al 1993]. The N-terminus domains share homologies with a consensus sequence of the whey acid protein family and a motif found in protease inhibitors. The C terminus contains a series of fibronectin type III repeats similar to those found in neural cell adhesion molecules.
Abnormal gene product. Impaired function of anosmin results in a migratory defect of the olfactory and GnRH neurons from the olfactory placode during development [Cariboni et al 2004]. The obstructed migration of these neurons accounts for the tell-tale signs of KS, IGD, and anosmia, and leads to olfactory bulb malformation detectable by MRI in the majority of individuals.
CHD7
Gene structure. CHD7 comprises 38 exons.
Pathogenic variants. Pathogenic variants of CHD7 resulting in IGD are predominantly missense variants that are either hypomorphic alleles or dominant alleles; in contrast, loss-of-function (i.e., truncating) pathogenic variants in CHD7 lead to a more extensive phenotypic presentation as seen in individuals with CHARGE syndrome [Balasubramanian et al 2014].
Normal gene product. The normal gene product is chromodomain helicase DNA-binding protein 7. It belongs to a family of proteins that are thought to alter nucleosome structures and mediate chromatin interactions.
Abnormal gene product. CHD7 pathogenic variants reported in individuals with KS or nIGD result in truncated proteins or amino acid substitutions of conserved residues when compared with CHD7 orthologs [Kim et al 2008].
FGFR1
Gene structure. FGFR1 comprises 18 exons with a known splice variant at the end of exon 10.
Pathogenic variants. Pathogenic variants in FGFR1 include pathogenic deletions and missense, nonsense, and splice variants. For more information, see Table A.
Normal gene product. FGFR1 encodes a membrane receptor with three extracellular immunoglobulin-like domains and an intracellular tyrosine kinase domain [Lee et al 1989]. Ligand binding results in receptor dimerization and recruitment of intracellular signaling proteins.
Abnormal gene product. Abnormal FGFR1 gene products result in impaired receptor signaling. The gene dose effect of anosmin and its interaction with FGFR1 in guiding GnRH neuronal migration have been proposed as explanations for the greater predominance of the IGD phenotype in males than females [Dodé et al 2003].
GNRHR
Gene structure. GNRHR comprises three coding exons and one alternative splice variant.
Pathogenic variants. GNRHR pathogenic variants, typically missense variants, cause normosmic IGD in an autosomal recessive inheritance pattern. Other pathogenic variants in GNRHR include nonsense and frameshift variants that occasionally cause autosomal recessive IGD. Heterozygous pathogenic variants (missense, frameshift, and nonsense) are also seen in persons with IGD with diverse clinical phenotypes, suggesting an oligogenic inheritance pattern [Gianetti et al 2012].
Normal gene product. GNRHR encodes for the gonadotropin-releasing hormone receptor, GNRHR, a G protein-coupled transmembrane receptor for the decapeptide, GNRH.
Abnormal gene product. Abnormal GNRHR gene products result in diminished or absent GNRH signaling through the receptor resulting in hypogonadotropism.
IL17RD
Gene structure. IL17RD comprises 17 coding exons and seven alternative splice variants.
Pathogenic variants. IL17RD missense variants in both homozygous and heterozygous state cause the KS form of IGD, including oligogenic inheritance.
Normal gene product. This gene encodes a membrane protein that belongs to the interleukin-17 receptor (IL-17R) protein family and is the component of the interleukin-17 receptor signaling complex. The gene product affects fibroblast growth factor signaling, inhibiting or stimulating growth through MAPK/ERK signaling.
Abnormal gene product. Abnormal IL17RD gene product results in increased FGF8 signaling in vitro, resulting in apoptosis of olfactory progenitor cells.
PROKR2
Gene structure. PROKR2 comprises two exons.
Pathogenic variants. Pathogenic variants of PROKR2 described include missense and nonsense variants.
Normal gene product. The normal gene product encodes the prokineticin receptor 2, a G protein-coupled transmembrane receptor for PROK2.
Abnormal gene product. The PROKR2 pathogenic variants identified in individuals with KS/nIGD result in diminished receptor function and impaired signaling [Cole et al 2008, Monnier et al 2009, Martin et al 2011]. Functional studies of selected PROKR2 pathogenic variants have failed to demonstrate a dominant negative effect. Knockout mice lack olfactory bulbs and have severe atrophy of the reproductive system related to the absence of gonadotropin-releasing hormone (Gnrh)-synthesizing neurons in the hypothalamus [Matsumoto et al 2006, Martin et al 2011].
SOX10
Gene structure. SOX10 comprises four coding exons and four alternative splice variants.
Pathogenic variants. SOX10 nonsense, frameshift, or missense variants cause the KS form of IGD in an autosomal dominant pattern with variable penetrance.
Normal gene product. SOX10 encodes a transcription factor, SRY (sex determining region Y)-box 10 which is involved in the regulation of neural crest and peripheral nervous system development.
Abnormal gene product. Abnormal SOX10 gene products resulting in transcriptional activity and Sox10-deficient mice show impaired development of olfactory ensheathing cells and impaired migration of GnRH neurons.
TACR3
Gene structure. TACR3 comprises five coding exons and no alternative splice variants.
Pathogenic variants. TACR3 nonsense, frameshift, or missense variants cause normosmic IGD in an autosomal recessive inheritance pattern. Occasionally, heterozygous pathogenic variants are also seen in persons with IGD, suggesting an oligogenic inheritance pattern.
Normal gene product. TACR3 encodes for TACR3, a G protein-coupled transmembrane receptor for neurokinin B.
Abnormal gene product. Abnormal TACR3 gene products result in diminished or absent neurokinin B signaling through the receptor, resulting in secondary GnRH deficiency and consequent hypogonadotropism.
Chapter Notes
Author History
Margaret Au, MBE, MS, CGC; Massachusetts General Hospital (2010-2013)
Ravikumar Balasubramanian, MD, PhD (2013-present)
Cassandra Buck, MS, CGC; Massachusetts General Hospital (2013-2017)
Marissa Caudill; University of Connecticut Health Center (2007-2010)
William F Crowley Jr, MD (2007-present)
J Carl Pallais, MD, MPH; Massachusetts General Hospital (2007-2013)
Nelly Pitteloud, MD; Massachusetts General Hospital (2007-2013)
Stephanie Seminara, MD; Massachusetts General Hospital (2007-2013)
Revision History
- 12 May 2022 (aa) Revision: encephalocraniocutaneous lipomatosis added to Genetically Related Disorders
- 2 March 2017 (ha) Comprehensive update posted live
- 18 July 2013 (me) Comprehensive update posted live
- 8 April 2010 (me) Comprehensive update posted live
- 23 May 2007 (me) Review posted live
- 1 June 2006 (jcp) Original submission
References
Published Guidelines / Consensus Statements
- Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Available online. 2015. Accessed 11-1-22.
Literature Cited
- Au MG, Crowley WF Jr, Buck CL. Genetic counseling for isolated GnRH deficiency. Mol Cell Endocrinol. 2011;346:102–9. [PMC free article: PMC3185214] [PubMed: 21664415]
- Balasubramanian R, Choi JH, Francescatto L, Willer J, Horton ER, Asimacopoulos EP, Stankovic KM, Plummer L, Buck CL, Quinton R, Nebesio TD, Mericq V, Merino PM, Meyer BF, Monies D, Gusella JF, Al Tassan N, Katsanis N, Crowley WF Jr. Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc Natl Acad Sci U S A. 2014;111:17953–8. [PMC free article: PMC4273325] [PubMed: 25472840]
- Balasubramanian R, Chew S, MacKinnon SE, Kang PB, Andrews C, Chan WM, Engle EC. Expanding the phenotypic spectrum and variability of endocrine abnormalities associated with TUBB3 E410K syndrome. J Clin Endocrinol Metab. 2015;100:E473–7. [PMC free article: PMC4333039] [PubMed: 25559402]
- Bédécarrats GY, Kaiser UB. Mutations in the human gonadotropin-releasing hormone receptor: insights into receptor biology and function. Semin Reprod Med. 2007;25:368–78. [PubMed: 17710733]
- Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5:569–76. [PMC free article: PMC2864719] [PubMed: 19707180]
- Bick D, Curry CJ, McGill JR, Schorderet DF, Bux RC, Moore CM. Male infant with ichthyosis, Kallmann syndrome, chondrodysplasia punctata, and an Xp chromosome deletion. Am J Med Genet. 1989;33:100–7. [PubMed: 2750777]
- Bin-Abbas B, Conte FA, Grumbach MM, Kaplan SL. Congenital hypogonadotropic hypogonadism and micropenis: effect of testosterone treatment on adult penile size why sex reversal is not indicated. J Pediatr. 1999;134:579–83. [PubMed: 10228293]
- Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11:547–64. [PubMed: 26194704]
- Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, Lombès M, Millar RP, Guiochon-Mantel A, Young J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–8. [PubMed: 19535795]
- Burris AS, Rodbard HW, Winters SJ, Sherins RJ. Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size. J Clin Endocrinol Metab. 1988;66:1144–51. [PubMed: 3372679]
- Cariboni A, Davidson K, Rakic S, Maggi R, Parnavelas JG, Ruhrberg C. Defective gonadotropin-releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: implications for the aetiology of hypogonadotropic hypogonadism. Hum Mol Genet. 2011;20:336–44. [PubMed: 21059704]
- Cariboni A, Pimpinelli F, Colamarino S, Zaninetti R, Piccolella M, Rumio C, Piva F, Rugarli EI, Maggi R. The product of X-linked Kallmann's syndrome gene (KAL1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum Mol Genet. 2004;13:2781–91. [PubMed: 15471890]
- Cariboni A, André V, Chauvet S, Cassatella D, Davidson K, Caramello A, Fantin A, Bouloux P, Mann F, Ruhrberg C. Dysfunctional SEMA3E signaling underlies gonadotropin-releasing hormone neuron deficiency in Kallmann syndrome. J Clin Invest. 2015;125:2413–28. [PMC free article: PMC4497752] [PubMed: 25985275]
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi Syndrome. Genet Med. 2012;14:10–26. [PubMed: 22237428]
- Cerrato F, Shagoury J, Kralickova M, Dwyer A, Falardeau J, Ozata M, Van Vliet G, Bouloux P, Hall JE, Hayes FJ, Pitteloud N, Martin KA, Welt C, Seminara SB. Coding sequence analysis of GNRHR and GPR54 in patients with congenital and adult-onset forms of hypogonadotropic hypogonadism. Eur J Endocrinol. 2006 Nov;155 Suppl 1:S3–S10. [PubMed: 17074994]
- Chan YM, Broder-Fingert S, Paraschos S, Lapatto R, Au M, Hughes V, Bianco SD, Min L, Plummer L, Cerrato F, De Guillebon A, Wu IH, Wahab F, Dwyer A, Kirsch S, Quinton R, Cheetham T, Ozata M, Ten S, Chanoine JP, Pitteloud N, Martin KA, Schiffmann R, Van der Kamp HJ, Nader S, Hall JE, Kaiser UB, Seminara SB. GnRH-deficient phenotypes in humans and mice with heterozygous variants in KISS1/Kiss1. J Clin Endocrinol Metab. 2011;96:E1771–81. [PMC free article: PMC3205899] [PubMed: 21880801]
- Chan YM, de Guillebon A, Lang-Muritano M, Plummer L, Cerrato F, Tsiaras S, Gaspert A, Lavoie HB, Wu CH, Crowley WF Jr, Amory JK, Pitteloud N, Seminara SB. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2009;106:11703–8. [PMC free article: PMC2710623] [PubMed: 19567835]
- Chew S, Balasubramanian R, Chan WM, Kang PB, Andrews C, Webb BD, MacKinnon SE, Oystreck DT, Rankin J, Crawford TO, Geraghty M, Pomeroy SL, Crowley WF Jr, Jabs EW, Hunter DG, Grant PE, Engle EC. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain. 2013;136:522–35. [PMC free article: PMC3572929] [PubMed: 23378218]
- Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougnères P, Lebouc Y, Froguel P, Guy-Grand B. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. [PubMed: 9537324]
- Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, Hughes VA, Dwyer AA, Ravio T, Hayes FJ, Seminara SB, Huot C, Alos N, Speiser P, Takeshita A, Van Vliet G, Pearce S, Crowley WF Jr, Zhou QY, Pitteloud N. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotropin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab. 2008;93:3551–9. [PMC free article: PMC2567850] [PubMed: 18559922]
- Costa-Barbosa FA, Balasubramanian R, Keefe KW, Shaw ND, Al-Tassan N, Plummer L, Dwyer A, Buck CL, Choi J-H, Seminara SB, Quinton R, Monies D, Meyer B, Hall JE, Pitteloud N, Crowley WF. prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab. 2013;98:E943–53. [PMC free article: PMC3644607] [PubMed: 23533228]
- Crowley WF Jr, Filicori M, Spratt DI, Santoro NF. The physiology of gonadotropin-releasing hormone (GnRH) secretion in men and women. Recent Prog Horm Res. 1985;41:473–531. [PubMed: 3931190]
- de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–6. [PMC free article: PMC196911] [PubMed: 12944565]
- de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337:1597–602. [PubMed: 9371856]
- Degros V, Cortet-Rudelli C, Soudan B, Dewailly D. The human chorionic gonadotropin test is more powerful than the gonadotropin-releasing hormone agonist test to discriminate male isolated hypogonadotropic hypogonadism from constitutional delayed puberty. Eur J Endocrinol. 2003;149:23–9. [PubMed: 12824862]
- Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–5. [PubMed: 12627230]
- Doty RL. Office procedures for quantitative assessment of olfactory function. Am J Rhinol. 2007;21:460–73. [PubMed: 17882917]
- Dwyer AA, Sykiotis GP, Hayes FJ, Boepple PA, Lee H, Loughlin KR, Dym M, Sluss PM, Crowley WF Jr, Pitteloud N. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2013;98:E1790–5. [PMC free article: PMC3816270] [PubMed: 24037890]
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–31. [PMC free article: PMC2441855] [PubMed: 18596921]
- Gianetti E, Hall JE, Au MG, Kaiser UB, Quinton R, Stewart JA, Metzger DL, Pitteloud N, Mericq V, Merino PM, Levitsky LL, Izatt L, Lang-Muritano M, Fujimoto VY, Dluhy RG, Chase ML, Crowley WF Jr, Plummer L, Seminara SB. When genetic load does not correlate with phenotypic spectrum: lessons from the GnRH receptor (GNRHR). J Clin Endocrinol Metab. 2012;97:E1798–807. [PMC free article: PMC3431570] [PubMed: 22745237]
- Gianetti E, Tusset C, Noel SD, Au MG, Dwyer AA, Hughes VA, Abreu AP, Carroll J, Trarbach E, Silveira LF, Costa EM, de Mendonça BB, de Castro M, Lofrano A, Hall JE, Bolu E, Ozata M, Quinton R, Amory JK, Stewart SE, Arlt W, Cole TR, Crowley WF, Kaiser UB, Latronico AC, Seminara SB. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 2010;95:2857–67. [PMC free article: PMC2902066] [PubMed: 20332248]
- Grumbach MM. A window of opportunity: the diagnosis of gonadotropin deficiency in the male infant. J Clin Endocrinol Metab. 2005;90:3122–7. [PubMed: 15728198]
- Guran T, Tolhurst G, Bereket A, Rocha N, Porter K, Turan S, Gribble FM, Kotan LD, Akcay T, Atay Z, Canan H, Serin A, O'Rahilly S, Reimann F, Semple RK, Topaloglu AK. Hypogonadotropic hypogonadism due to a novel missense mutation in the first extracellular loop of the neurokinin B receptor. J Clin Endocrinol Metab. 2009;94:3633–9. [PMC free article: PMC4306717] [PubMed: 19755480]
- Hanchate NK, Giacobini P, Lhuillier P, Parkash J, Espy C, Fouveaut C, Leroy C, Baron S, Campagne C, Vanacker C, Collier F, Cruaud C, Meyer V, García-Piñero A, Dewailly D, Cortet-Rudelli C, Gersak K, Metz C, Chabrier G, Pugeat M, Young J, Hardelin JP, Prevot V, Dodé C. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 2012;8:e1002896. [PMC free article: PMC3426548] [PubMed: 22927827]
- Hipkin LJ, Casson IF, Davis JC. Identical twins discordant for Kallmann's syndrome. J Med Genet. 1990;27:198–9. [PMC free article: PMC1017005] [PubMed: 2325096]
- Hoffman AR, Crowley WF Jr. Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 1982;307:1237–41. [PubMed: 6813732]
- Hou JW. Detection of gene deletions in children with chondrodysplasia punctata, ichthyosis, Kallmann syndrome, and ocular albinism by FISH studies. Chang Gung Med J. 2005;28:643–50. [PubMed: 16323556]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Hutchins BI, Kotan LD, Taylor-Burds C, Ozkan Y, Cheng PJ, Gurbuz F, Tiong JD, Mengen E, Yuksel B, Topaloglu AK, Wray S. CCDC141 Mutation Identified in Anosmic Hypogonadotropic Hypogonadism (Kallmann Syndrome) Alters GnRH Neuronal Migration. Endocrinology. 2016;157:1956–66. [PMC free article: PMC4870868] [PubMed: 27014940]
- Jackson RS, Creemers JW, Farooqi IS, Raffin-Sanson ML, Varro A, Dockray GJ, Holst JJ, Brubaker PL, Corvol P, Polonsky KS, Ostrega D, Becker KL, Bertagna X, Hutton JC, White A, Dattani MT, Hussain K, Middleton SJ, Nicole TM, Milla PJ, Lindley KJ, O'Rahilly S. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. 2003;112:1550–60. [PMC free article: PMC259128] [PubMed: 14617756]
- Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O'Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–6. [PubMed: 9207799]
- Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, van der Donk K, Seminara S, Bergman JE, Brunner HG, Crowley WF Jr, Hoefsloot LH. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65–71. [PMC free article: PMC2854009] [PubMed: 19021638]
- Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, Ha KS, Itokawa Y, Meliciani I, Wenzel W, Lee D, Rosenberger G, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2010;87:465–79. [PMC free article: PMC2948809] [PubMed: 20887964]
- Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83:511–9. [PMC free article: PMC2561938] [PubMed: 18834967]
- Kotan LD, Hutchins BI, Ozkan Y, Demirel F, Stoner H, Cheng PJ, Esen I, Gurbuz F, Bicakci YK, Mengen E, Yuksel B, Wray S, Topaloglu AK. Mutations in FEZF1 cause Kallmann syndrome. Am J Hum Genet. 2014;95:326–31. [PMC free article: PMC4157145] [PubMed: 25192046]
- Kotan LD, Cooper C, Darcan Ş, Carr IM, Özen S, Yan Y, Hamedani MK, Gürbüz F, Mengen E, Turan İ, Ulubay A, Akkuş G, Yüksel B, Topaloğlu AK, Leygue E. Idiopathic Hypogonadotropic Hypogonadism Caused by Inactivating Mutations in SRA1. J Clin Res Pediatr Endocrinol. 2016;8:125–34. [PMC free article: PMC5096466] [PubMed: 27086651]
- Laitinen EM, Vaaralahti K, Tommiska J, Eklund E, Tervaniemi M, Valanne L, Raivio T. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J Rare Dis. 2011;6:41. [PMC free article: PMC3143089] [PubMed: 21682876]
- Lalani SR, Belmont JW. CHARGE syndrome. In: Lang F, ed. Encyclopedia of Molecular Mechanisms of Disease. Springer Publishing House. 2009.
- Lee PL, Johnson DE, Cousens LS, Fried VA, Williams LT. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science. 1989;245:57–60. [PubMed: 2544996]
- Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L, Au MG, Doty RL, Seminara SB, Chan YM, Pitteloud N, Crowley WF Jr, Balasubramanian R. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J Clin Endocrinol Metab. 2012;97:E136–44. [PMC free article: PMC3251934] [PubMed: 22072740]
- Liu PY, Baker HW, Jayadev V, Zacharin M, Conway AJ, Handelsman DJ. Induction of spermatogenesis and fertility during gonadotropin treatment of gonadotropin-deficient infertile men: predictors of fertility outcome. J Clin Endocrinol Metab. 2009;94:801–8. [PubMed: 19066302]
- Mainieri AS, Elnecave RH. Usefulness of the free alpha-subunit to diagnose hypogonadotropic hypogonadism. Clin Endocrinol (Oxf). 2003;59:307–13. [PubMed: 12919153]
- Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, Hall JE, Adam I, Dwyer A, Plummer L, Aldrin SV, O'Rourke J, Kirby A, Lage K, Milunsky A, Milunsky JM, Chan J, Hedley-Whyte ET, Daly MJ, Katsanis N, Seminara SB. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368:1992–2003. [PMC free article: PMC3738065] [PubMed: 23656588]
- Martin C, Balasubramanian R, Dwyer AA, Au MG, Sidis Y, Kaiser UB, Seminara SB, Pitteloud N, Zhou QY, Crowley WF Jr. The Role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr Rev. 2011;32:225–46. [PMC free article: PMC3365793] [PubMed: 21037178]
- Martin K, Santoro N, Hall J, Filicori M, Wierman M, Crowley WF Jr (1990) Clinical review 15: Management of ovulatory disorders with pulsatile gonadotropin-releasing hormone. J Clin Endocrinol Metab. 71:1081A-G. [PubMed: 2229271]
- Massin N, Pecheux C, Eloit C, Bensimon JL, Galey J, Kuttenn F, Hardelin JP, Dode C, Touraine P. X chromosome-linked Kallmann syndrome: clinical heterogeneity in three siblings carrying an intragenic deletion of the KAL-1 gene. J Clin Endocrinol Metab. 2003;88:2003–8. [PubMed: 12727945]
- Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, Hiyama H, Matsumoto M, Takasaki J, Kamohara M, Matsuo A, Ishii H, Kobori M, Katoh M, Matsushime H, Furuichi K, Shigeyoshi Y. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci U S A. 2006;103:4140–5. [PMC free article: PMC1449660] [PubMed: 16537498]
- Matsuo T, Okamoto S, Izumi Y, Hosokawa A, Takegawa T, Fukui H, Tun Z, Honda K, Matoba R, Tasumi K, Amino N. A novel mutation of the KAL1 gene in monozygotic twins with Kallmann syndrome. Eur J Endocrinol. 2000;143:783–7. [PubMed: 11124862]
- Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, Beenken A, Clarke J, Pers TH, Dworzynski P, Keefe K, Niedziela M, Raivio T, Crowley WF Jr, Seminara SB, Quinton R, Hughes VA, Kumanov P, Young J, Yialamas MA, Hall JE, Van Vliet G, Chanoine JP, Rubenstein J, Mohammadi M, Tsai PS, Sidis Y, Lage K, Pitteloud N. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet. 2013;92:725–43. [PMC free article: PMC3644636] [PubMed: 23643382]
- Monnier C, Dode C, Fabre L, Teixeira L, Labesse G, Pin JP, Hardelin JP, Rondard P. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum Mol Genet. 2009;18:75–81. [PMC free article: PMC3298864] [PubMed: 18826963]
- Nachtigall LB, Boepple PA, Pralong FP, Crowley WF Jr. Adult-onset idiopathic hypogonadotropic hypogonadism--a treatable form of male infertility. N Engl J Med. 1997;336:410–5. [PubMed: 9010147]
- Oliveira LM, Seminara SB, Beranova M, Hayes FJ, Valkenburgh SB, Schipani E, Costa EM, Latronico AC, Crowley WF Jr, Vallejo M. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. 2001;86:1532–8. [PubMed: 11297579]
- Pedersen-White JR, Chorich LP, Bick DP, Sherins RJ, Layman LC. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Hum Reprod. 2008;14:367–70. [PMC free article: PMC2434956] [PubMed: 18463157]
- Pingault V, Bodereau V, Baral V, Marcos S, Watanabe Y, Chaoui A, Fouveaut C, Leroy C, Vérier-Mine O, Francannet C, Dupin-Deguine D, Archambeaud F, Kurtz FJ, Young J, Bertherat J, Marlin S, Goossens M, Hardelin JP, Dodé C, Bondurand N. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am J Hum Genet. 2013;92:707–24. [PMC free article: PMC3644631] [PubMed: 23643381]
- Pinto G, Abadie V, Mesnage R, Blustajn J, Cabrol S, Amiel J, Hertz-Pannier L, Bertrand AM, Lyonnet S, Rappaport R, Netchine I. CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab. 2005;90:5621–6. [PubMed: 16030162]
- Pitteloud N, Acierno JS Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, Grant E, Mohammadi M, Crowley WF Jr. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2006a;103:6281–6. [PMC free article: PMC1458869] [PubMed: 16606836]
- Pitteloud N, Boepple PA, DeCruz S, Valkenburgh SB, Crowley WF Jr, Hayes FJ. The fertile eunuch variant of idiopathic hypogonadotropic hypogonadism: spontaneous reversal associated with a homozygous mutation in the gonadotropin-releasing hormone receptor. J Clin Endocrinol Metab. 2001;86:2470–5. [PubMed: 11397842]
- Pitteloud N, Hayes FJ, Boepple PA, DeCruz S, Seminara SB, MacLaughlin DT, Crowley WF Jr. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002a;87:152–60. [PubMed: 11788640]
- Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF Jr. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002b;87:4128–36. [PubMed: 12213860]
- Pitteloud N, Meysing A, Quinton R, Acierno JS Jr, Dwyer AA, Plummer L, Fliers E, Boepple P, Hayes F, Seminara S, Hughes VA, Ma J, Bouloux P, Mohammadi M, Crowley WF Jr. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 2006b;254-255:60–9. [PubMed: 16764984]
- Quinton R, Duke VM, Robertson A, Kirk JM, Matfin G, de Zoysa PA, Azcona C, MacColl GS, Jacobs HS, Conway GS, Besser M, Stanhope RG, Bouloux PM. Idiopathic gonadotropin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf). 2001;55:163–74. [PubMed: 11531922]
- Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, Cole LW, Pearce SH, Lee H, Boepple P, Crowley WF Jr, Pitteloud N. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357:863–73. [PubMed: 17761590]
- Richards MR, Plummer L, Chan YM, Lippincott MF, Quinton R, Kumanov P, Seminara SB. Phenotypic spectrum of POLR3B mutations: isolated hypogonadotropic hypogonadism without neurological or dental anomalies. J Med Genet. 2017;54:19–25. [PMC free article: PMC5189673] [PubMed: 27512013]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rugarli EI, Lutz B, Kuratani SC, Wawersik S, Borsani G, Ballabio A, Eichele G. Expression pattern of the Kallmann syndrome gene in the olfactory system suggests a role in neuronal targeting. Nat Genet. 1993;4:19–26. [PubMed: 8513320]
- Salian-Mehta S, Xu M, Knox AJ, Plummer L, Slavov D, Taylor M, Bevers S, Hodges RS, Crowley WF Jr, Wierman ME. Functional consequences of AXL sequence variants in hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2014;99:1452–60. [PMC free article: PMC3973777] [PubMed: 24476074]
- Santoro N, Filicori M, Crowley WF Jr. Hypogonadotropic disorders in men and women: diagnosis and therapy with pulsatile gonadotropin-releasing hormone. Endocr Rev. 1986;7:11–23. [PubMed: 3082615]
- Sato N, Ogata T. Kallmann syndrome. Nihon Rinsho. 2006 Suppl 2:220–4. [PubMed: 16817388]
- Segal TY, Mehta A, Anazodo A, Hindmarsh PC, Dattani MT. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty. J Clin Endocrinol Metab. 2009;94:780–5. [PubMed: 19017752]
- Seminara SB, Hayes FJ, Crowley WF Jr. Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations. Endocr Rev. 1998;19:521–39. [PubMed: 9793755]
- Seminara SB, Acierno JS Jr, Abdulwahid NA, Crowley WF Jr, Margolin DH. Hypogonadotropic hypogonadism and cerebellar ataxia: detailed phenotypic characterization of a large, extended kindred. J Clin Endocrinol Metab. 2002;87:1607–12. [PubMed: 11932290]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MB, Crowley WF Jr, Aparicio SA, Colledge WH. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–27. [PubMed: 14573733]
- Semple RK, Achermann JC, Ellery J, Farooqi IS, Karet FE, Stanhope RG, O'rahilly S, Aparicio SA. Two novel missense mutations in g protein-coupled receptor 54 in a patient with hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2005;90:1849–55. [PubMed: 15598687]
- Shaw ND, Seminara SB, Welt CK, Au MG, Plummer L, Hughes VA, Dwyer AA, Martin KA, Quinton R, Meriq V, Merino PM, Gusella JF, Crowley WF Jr, Pitteloud N, Hall JE. Expanding the phenotype and genotype of female GnRH deficiency. J. Clin. Endocrinol. Metab. 2011;96:E566–76. [PMC free article: PMC3047229] [PubMed: 21209029]
- Sidhoum VF, Chan YM, Lippincott MF, Balasubramanian R, Quinton R, Plummer L, Dwyer A, Pitteloud N, Hayes FJ, Hall JE, Martin KA, Boepple PA, Seminara SB. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J Clin Endocrinol Metab. 2014;99:861–70. [PMC free article: PMC3942233] [PubMed: 24423288]
- Spratt DI, Carr DB, Merriam GR, Scully RE, Rao PN, Crowley WF Jr. The spectrum of abnormal patterns of gonadotropin-releasing hormone secretion in men with idiopathic hypogonadotropic hypogonadism: clinical and laboratory correlations. J Clin Endocrinol Metab. 1987;64:283–91. [PubMed: 3098771]
- Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–5. [PubMed: 9500540]
- Suzuki E, Izumi Y, Chiba Y, Horikawa R, Matsubara Y, Tanaka M, Ogata T, Fukami M, Naiki Y. Loss-of-function SOX10 mutation in a patient with Kallmann syndrome, hearing loss, and iris hypopigmentation. Horm Res Paediatr. 2015;84:212–6. [PubMed: 26228106]
- Sykiotis GP, Hoang XH, Avbelj M, Hayes FJ, Thambundit A, Dwyer A, Au M, Plummer L, Crowley WF Jr, Pitteloud N. Congenital idiopathic hypogonadotropic hypogonadism: evidence of defects in the hypothalamus, pituitary, and testes. J Clin Endocrinol Metab. 2010a;95:3019–27. [PMC free article: PMC2902061] [PubMed: 20382682]
- Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, Dwyer AA, Quinton R, Hall JE, Gusella JF, Seminara SB, Crowley WF Jr, Pitteloud N. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A. 2010b;107:15140–4. [PMC free article: PMC2930591] [PubMed: 20696889]
- Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Ozbek MN, Imamoglu S, Akalin NS, Yuksel B, O'Rahilly S, Semple RK. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354–8. [PMC free article: PMC4312696] [PubMed: 19079066]
- Topaloglu AK, Tello JA, Kotan LD, Ozbek MN, Yilmaz MB, Erdogan S, Gurbuz F, Temiz F, Millar R, Yuksel B. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. NEJM. 2012;366:629–35. [PubMed: 22335740]
- Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, Quinton R, Hughes V, Seminara SB, Van Uum S, Crowley WF, Habuchi H, Kimataf K, Piteloud N, Bülow HE. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2011;108:11524–9. [PMC free article: PMC3136273] [PubMed: 21700882]
- Trarbach EB, Abreu AP, Silveira LF, Garmes HM, Baptista MT, Teles MG, Costa EM, Mohammadi M, Pitteloud N, de Mendonca BB, Latronico AC. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. J Clin Endocrinol Metab. 2010a;95:3491–6. [PMC free article: PMC3213864] [PubMed: 20463092]
- Trarbach EB, Teles MG, Costa EM, Abreu AP, Garmes HM, Guerra G Jr, Baptista MT, de Castro M, Mendonca BB, Latronico AC. Screening of autosomal gene deletions in patients with hypogonadotropic hypogonadism using multiplex ligation-dependent probe amplification: detection of a hemizygosis for the fibroblast growth factor receptor 1. Clin Endocrinol (Oxf). 2010b;72:371–6. [PubMed: 19489874]
- Van Dop C, Burstein S, Conte FA, Grumbach MM. Isolated gonadotropin deficiency in boys: clinical characteristics and growth. J Pediatr. 1987;111:684–92. [PubMed: 2889818]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–7. [PubMed: 15300250]
- Waldhauser F, Weissenbacher G, Frisch H, Pollak A. Pulsatile secretion of gonadotropins in early infancy. Eur J Pediatr. 1981;137:71–4. [PubMed: 6791927]
- Waldstreicher J, Seminara SB, Jameson JL, Geyer A, Nachtigall LB, Boepple PA, Holmes LB, Crowley WF Jr. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab. 1996;81:4388–95. [PubMed: 8954047]
- Whitcomb RW, Crowley WF Jr. Clinical review 4: Diagnosis and treatment of isolated gonadotropin-releasing hormone deficiency in men. J Clin Endocrinol Metab. 1990;70:3–7. [PubMed: 2403572]
- Xu N, Kim HG, Bhagavath B, Cho SG, Lee JH, Ha K, Meliciani I, Wenzel W, Podolsky RH, Chorich LP, Stackhouse KA, Grove AM, Odom LN, Ozata M, Bick DP, Sherins RJ, Kim SH, Cameron RS, Layman LC. Nasal embryonic LHRH factor (NELF) mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil Steril. 2011;95:1613–20.e1. [PMC free article: PMC3888818] [PubMed: 21300340]
- Young J. Approach to the male patient with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2012;97:707–18. [PubMed: 22392951]
Publication Details
Author Information and Affiliations
Reproductive Endocrine Unit
Massachusetts General Hospital
Boston, Massachusetts
Chief, Reproductive Endocrine Unit of the Department of Medicine
Massachusetts General Hospital
Boston, Massachusetts
Publication History
Initial Posting: May 23, 2007; Last Revision: May 12, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Balasubramanian R, Crowley WF Jr. Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency. 2007 May 23 [Updated 2022 May 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.