Clinical Description
Classic Schinzel-Giedion syndrome (SGS), an ultra-rare multisystem disorder, is characterized by a range of physical and developmental abnormalities. The main features include global neurodevelopmental impairment leading to moderate-to-profound intellectual disability, epilepsy (often refractory to treatment), tone abnormalities, dysautonomia, and cerebral visual and hearing impairment. Poor weight gain is common and often associated with gastroesophageal reflux disease, chronic vomiting, constipation, gastroparesis, and/or feeding intolerance. Structural malformations can involve the heart, skeleton, kidney and urinary tract, genitalia, and brain. Rarely there may be anomalies of the liver, spleen, and/or pancreas. Other features may include neuroepithelial neoplasia, severely disrupted sleep, choanal stenosis, inguinal hernia, sensitive skin, and increased risk of infection.
To date, more than 50 individuals have been reported with molecularly confirmed classic SGS [Hoischen et al 2010, Suphapeetiporn et al 2011, Lestner et al 2012, Ko et al 2013, Carvalho et al 2015, Herenger et al 2015, López-González et al 2015, Miyake et al 2015, Takeuchi et al 2015, Volk et al 2015, Hishimura et al 2016, Acuna-Hidalgo et al 2017, Bulut et al 2017, Leonardi et al 2020, Leone et al 2020, Sullivan et al 2020, Yang et al 2022].
In addition, more than 40 individuals with the clinical diagnosis of SGS were reported in the medical literature before pathogenic variants in SETBP1 were identified to cause SGS [Lehman et al 2008].
Atypical SGS, caused by SETBP1 pathogenic variants in proximity to – but not within – the mutational hot spot, has a broad spectrum of clinical features of variable severity that partially overlap with classic SGS, but do not include risk for neuroepithelial neoplasia to date. Five individuals with atypical SGS have been reported to date.
The following clinical description is mainly based on reports of individuals with molecularly confirmed classic SGS (see Table 3).
Table 3.
Classic Schinzel-Giedion Syndrome: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature |
|---|
| Development delay / intellectual disability |
100%
|
| Neurologic | Epilepsy | 75%-100% |
| Hypotonia | 75%-100% |
| Cerebral visual impairment | 70%-80%% |
| Spasticity/hypertonia | 50%-75% |
| Feeding difficulties & poor weight gain | 75%-100% |
| Hearing impairment | 75%-100% |
| Ophthalmologic involvement | 75%-100% |
| Skeletal findings | 75%-100% |
| CAKUT | 75%-100% |
| Genital anomalies | 75%-100% |
| Hypertrichosis | 50%-75% |
| Cardiac defects | 20%-50% |
| Neoplasia | 20%-50% |
| Tracheo- & laryngomalacia | 20%-50% |
| Choanal stenosis | 20%-50% |
| Inguinal hernia | 20%-50% |
| Frequent infections | 20%-50% |
CAKUT = congenital anomalies of the kidney and urinary tract
Developmental delay (DD) and intellectual disability (ID). Moderate-to-severe (in classic SGS) or mild-to-severe (in atypical SGS) developmental delay and intellectual disability are present in all individuals reported to date. The majority of individuals with classic SGS have no speech and never develop the ability to walk independently.
Epilepsy may start in the neonatal period or later. The heterogeneous seizure types include tonic, tonic-clonic, myoclonic, or partial motor seizures and infantile epileptic spasms syndrome (IESS) (seen in about 25% of individuals). EEG findings often show multifocal spikes or hypsarrhythmia. Epilepsy is reported less frequently in individuals with atypical SGS than in individuals with classic SGS.
Most seizures are refractory to treatment with anti-seizure medications (ASMs), adrenocorticotropic hormone, steroids, and the ketogenic diet.
Hypotonia and spasticity are common and may occur at different times in the same individual. While hypotonia is common at birth, spasticity may develop at a later age and progress. Differences in muscular tone may also lead to spinal deformities such as scoliosis.
Cerebral vision impairment has been reported in 70%-80% of individuals with classic SGS and may also occur in atypical SGS.
Feeding difficulties may be due to underlying factors including hypotonia, sucking problems due to micrognathia, swallowing problems, trachea- and/or laryngomalacia, gastroesophageal reflux disease, and/or vomiting.
Hearing impairment occurs in nearly 90% of individuals with classic SGS and may also occur in atypical SGS. While the type of hearing loss can vary among affected individuals, bilateral sensorineural hearing loss is most common, ranges from mild to profound, and can differ in each ear.
Other reported types of hearing loss include mixed or conductive hearing loss. For example, mixed moderate hearing loss was reported in two individuals due to bilateral deformations of the stapes (that had a tuning fork shape) and in one individual with a flattened last cochlear spire (identified by temporal bone CT scan) [Minn et al 2002, Herenger et al 2015].
Ophthalmologic involvement. Abnormalities of the optic nerve (small optic discs, optic disc pit), strabismus, and alacrima with corneal hypoesthesia have been reported.
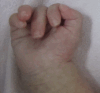
Skeletal findings. Anomalies identified on skeletal radiographs can include sclerotic base of the skull with a wide occipital synchondrosis, a poorly mineralized rest of the cranium, a wide anterior fontanelle, and wormian bones. Additional findings can include broad ribs, scoliosis, bowed long bones, short pubic rami, wide pubic symphysis, and hypoplastic distal phalanges in the hands and feet. Individuals often show a typical posture with clenched hand (see ). Talipes equinovarus is common.
Characteristic hand posture with clenched hand in an individual with Schinzel-Giedion syndrome Adapted with permission from Acuna-Hidalgo et al [2017]
Postaxial polydactyly (predominantly of the hands) was noted in about 5%-10% of individuals with classic SGS.
Congenital anomalies of the kidney and urinary tract. Bilateral or unilateral hydronephrosis, ranging from mild pyelectasis to severe hydronephrosis, is common in individuals with classic SGS. Hydronephrosis may be present on prenatal ultrasound examination; however, it may not be present at birth but rather develop during infancy. Two individuals with atypical SGS have had medullary cystic kidneys, mild pyelectasis, and chronic urinary infections.
Stenosis of the ureteropelvic junction or vesicoureteral reflux are common. Bladder atony may be the cause of frequent and persistent urinary tract infections.
Other anomalies can include abnormal ureters and renal cysts.
One individual had bilateral large coralliform (i.e., resembling the calyx cavities) renal stones that were predominantly calcium oxalate and calcium phosphate without pyelic dilatation [Herenger et al 2015].
Genital anomalies. The majority of individuals with classic SGS have genital anomalies. In males, these include micropenis (which may appear as ambiguous genitalia on antenatal ultrasound examination), hypospadias, hypoplastic scrotum, and cryptorchidism. In females, these include bifid uterus, hypoplastic uterus, hypoplastic labia majora or minora, and deep labial sulcus. The perineum is often short with an anteriorly displaced anus [Lehman et al 2008].
Cardiac defects. The majority of cardiac defects are atrial septal defects, persistent foramen ovale, and patent ductus arteriosus. Other cardiac findings that occur occasionally include hypoplasia of ventricles and cardiac hypertrophy.
Neoplasia. About 25% of individuals with classic SGS develop a neoplasia, often of neuroepithelial origin. To date, SGS-related neoplasia has not been reported in individuals with atypical SGS caused by SETBP1 pathogenic variants outside of the mutational hot spot. Neoplasias may be detected at birth, during the first year of life, or later in life (e.g., malignant degeneration of a multicystic dysplastic kidney) [Matsumoto et al 2005].
Sacrococcygeal teratoma is the most common tumor. Other reported tumors include hepatoblastoma, lumbosacral teratoma, lumbosacral primitive neuroectodermal tumor, Wilms tumor, and extradural ependymomas. To date, juvenile myelomonocytic leukemia has been reported in one individual [Acuna-Hidalgo et al 2017].
Although most children with SGS die from other causes, a 24-month-old child died of organ failure due to a sacrococcygeal teratoma and a five-year-old child died due to relapse of an extradural ependymal tumor [Acuna-Hidalgo et al 2017].
Other findings
Microcephaly has been observed in approximately 80% of individuals with classic SGS. In most reported individuals, microcephaly is postnatal – although the occipital-frontal circumference may be within normal limits at birth (almost always below the 50th and often near the 10th centile), head growth decelerates and microcephaly develops during infancy.
Structural brain abnormalities vary. Most common are partial or complete agenesis of the corpus callosum. Other findings can include progressive cortical atrophy, ventricular anomalies, hydrocephalus, cortical abnormalities, delayed myelination, and choroid plexus cysts.
In one individual, an abnormal posterior fossa with stretching of the pituitary stalk resulted in central diabetes insipidus and central hypothyroidism [
Santos et al 1994].
Respiratory abnormalities. Structural abnormalities that may lead to breathing and swallowing difficulties include choanal stenosis, micrognathia, tracheobronchomalacia, and lung hypoplasia. Difficulty managing oral and respiratory secretions resulting from progressive gingival hypertrophy and/or excessive mucus production may be additional factors that increase the risk of aspiration and respiratory infections including pneumonia.
Gastrointestinal problems. Constipation, gastroesophageal reflux disease, and aspiration are common. Chronic vomiting and gastroparesis may also occur. Anteriorly placed anus is often associated with genital anomalies in males and females.
Structural anomalies of internal organs, such as hypoplasia of the pancreatic tail, annular pancreas, splenopancreatic fusion, and hepatosplenomegaly have been reported.
Postmortem microscopic evaluation of the pancreas in a four-day-old infant showed dilated interlobular ducts, filled with eosinic mucus and surrounded by abundant connective tissue. Additionally, dilated glands and mucus depositions were found in larynx and bronchial glands; although these findings were like those observed in cystic fibrosis,
CFTR testing was normal [
Acuna Hidalgo et al 2017].
Skin. Generalized hypertrichosis that is common at birth may recede during infancy. Facial hemangiomata are present in 25% of individuals with classic SGS. Other features may include dry erythematous skin, redundant nuchal skin, cutis marmorata, hypoplastic nipples, hyperconvex nails, and hypoplastic dermal ridges.
Dental findings include hypodontia, delayed teeth eruption, and congenital thickened gingiva (unrelated to use of ASMs).
One infant developed gingival hyperplasia at age seven months (two weeks prior to the initiation of ASMs) that became severe after age two years. His gingivae were so large that they protruded from his mouth, pressed his tongue to the pharynx, and covered his teeth. Because of difficulty with eating and breathing, he underwent full mouth gingivectomy twice, once at age four years and again at age six years. Histologic findings were gingival fibrous hyperplasia with mucoid depositions [
Kondoh et al 2001]. Similar microscopic histologic findings were noted on postmortem examination in a four-day-old infant [
Acuna-Hidalgo et al 2017].
Neurobehavioral manifestations. To date, extensive information on behavior is not available. Although sleep disturbances and extensive periods of crying and irritability have been reported, additional evaluation is warranted for an underlying cause such as urinary tract infection and development of hydrocephalus or sacrococcygeal tumor.
Growth. Birth length and weight are often within normal limits.
Prenatal findings. Pregnancy may be complicated by polyhydramnios. Fetal ultrasound examination may reveal other findings such as hydronephrosis, ambiguous genitalia, cardiac defects, overlapping fingers, and/or a typical facial appearance.
Prognosis. The shortened life span of children with SGS may be correlated with the number and severity of the features present. While death mainly results from pneumonia (also after aspiration), other reported causes of death in early infancy include sepsis, lung hypoplasia, intractable epilepsy, and sudden cardiac arrest.