Summary
Clinical characteristics.
Schmid metaphyseal chondrodysplasia (SMCD) is characterized by progressive short stature that develops by age two years. The clinical and radiographic features are usually not present at birth, but manifest in early childhood with short limbs, genu varum, and waddling gait. Facial features and head size are normal. Radiographs show metaphyseal irregularities of the long bones (e.g., splaying, flaring, cupping); shortening of the tubular bones; widened growth plates; coxa vara; and anterior cupping, sclerosis, and splaying of the ribs. Mild hand involvement often includes shortening of the tubular bones and metaphyseal cupping of the metacarpals and proximal phalanges. Platyspondyly and vertebral end-plate irregularities are less common. Hand and vertebral involvement can resolve with age. Early motor milestones may be delayed due to orthopedic complications. Intelligence is normal. Joint pain in the knees and hips is common and may limit physical activity. Adult height is typically more than 3.5 SD below the mean, although a wide spectrum that overlaps normal height has been reported. There are no extraskeletal manifestations.
Diagnosis/testing.
The diagnosis of SMCD is established in a proband with characteristic clinical and radiographic features and/or identification of a heterozygous pathogenic variant in COL10A1 by molecular genetic testing.
Management.
Treatment of manifestations: Management of orthopedic complications by orthopedist, physiotherapist, occupational therapist, and pain specialist as indicated. Joint-friendly exercise, weight management; mobility device as needed; corrective osteotomy by guided growth surgery or valgus osteotomy may be considered in late childhood / adolescence in those with progressive or symptomatic varus deformity, significant coxa vara, triangular fragment in the interior femoral neck, or poor or deteriorating function; exercise and support from nutritionist to maintain healthy weight; psychosocial support; environmental or occupational modifications as needed for short stature with recommendations from occupational therapy as needed.
Surveillance: Annual growth assessment, clinical evaluation for orthopedic manifestations, and psychosocial evaluation.
Agents/circumstances to avoid: Obesity; physical activities that cause excessive joint strain.
Genetic counseling.
SMCD is inherited in an autosomal dominant manner. Most individuals diagnosed with SMCD have the disorder as the result of a de novo COL10A1 pathogenic variant. Some individuals diagnosed with SMCD have an affected parent. Each child of an individual with SMCD has a 50% chance of inheriting the COL10A1 pathogenic variant. If the proband and the proband's reproductive partner are affected with different dominantly inherited skeletal dysplasias, genetic counseling becomes more complicated because of the risk of inheriting two dominantly inherited bone growth disorders. If the COL10A1 pathogenic variant has been identified in the affected parent, prenatal testing for a pregnancy at increased risk for SMCD and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
No formal diagnostic criteria for Schmid metaphyseal chondrodysplasia (SMCD) have been established. The diagnosis should be suspected in individuals with the following clinical, laboratory, and radiographic findings.
Clinical findings
- Short-limbed short stature by age two years (in >60%)
- Genu varum (bowed legs) (>60%)
- Waddling gait (>80%)
- Lumbar lordosis by age three to five years
- Normal craniofacies and absence of extraskeletal manifestations
Laboratory findings. Normal serum calcium, phosphate, vitamin D, and alkaline phosphatase
Radiographic findings (see Figure 1)
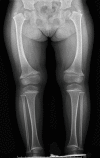
- Shortening of the tubular bones (>60%)
- Metaphyseal irregularities of the long bones (e.g., splaying, flaring, cupping) especially the proximal and distal femora (~100%)
- Widening of the growth plates
- Coxa vara (>80%)
- Anterior cupping, sclerosis, and splaying of the ribs (>90%)
- Mild hand involvement including shortening of the tubular bones and metaphyseal cupping of the metacarpals and proximal phalanges (~50%). Radiographic phalangeal and metacarpal findings may resolve with age.
- Vertebral involvement including platyspondyly and end-plate irregularities (~10%)
Establishing the Diagnosis
The diagnosis of SMCD is established in a proband with characteristic clinical and radiographic features and/or a heterozygous pathogenic variant in COL10A1 identified by molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype and consideration of alternate diagnoses.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of SMCD is broad and overlaps with other metaphyseal chondrodysplasias, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype overlapping with other inherited disorders with metaphyseal dysplasia are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and radiographic findings suggest a diagnosis of SMCD, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Perform sequence analysis of COL10A1 to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.Note: All pathogenic variants reported to date are missense variants in COL10A1; thus, testing for deletion or duplication is expected to be of very low yield.
- A multigene panel that includes COL10A1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the phenotype overlaps other inherited disorders characterized by metaphyseal dysplasia, comprehensive genomic testing is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Schmid Metaphyseal Chondrodysplasia
Clinical Characteristics
Clinical Description
Schmid metaphyseal chondrodysplasia (SMCD) is typically diagnosed in early childhood and is the most common and least severe metaphyseal chondrodysplasia [Al Kaissi et al 2018]. It results from disrupted calcification of metaphyseal cartilage and consequent restricted longitudinal growth of bones with preservation of the epiphyses. The clinical and radiographic features are usually not present at birth, but manifest in early childhood with limb shortening, genu varum, and a waddling gait [Bateman et al 2005]. The pattern of radiographic features is generally similar across individuals with SMCD, but clinical severity varies considerably [Mäkitie et al 2005]. There are no extraskeletal manifestations.
A comprehensive review of the published reports identified at least 130 unrelated individuals with SMCD and a confirmed pathogenic variant in COL10A1. The following description of the phenotypic features associated with this condition is based on these reports and author observations in a multidisciplinary skeletal dysplasia clinic.
Table 2.
Clinical and Radiographic Features of Schmid Metaphyseal Chondrodysplasia
Growth. Most neonates with SMCD have normal growth parameters. Progressive growth failure typically begins in the second year of life with individuals coming to medical attention after age two years with short-limbed short stature and bowed legs [Mäkitie et al 2005]. Adult height is typically more than 3.5 SD below the mean, although a wide spectrum that overlaps normal height has been reported [Mäkitie et al 2005]. No standard growth curves for SMCD are available.
Musculoskeletal. Most individuals with SMCD have genu varum (outward bowing at the knees), although valgus deformity has been reported. Waddling gait due to coxa vara is common by age two years and may require surgical correction. Joint pain in the knees and hips is common in adults and children with the condition and may limit physical activity. Chronic joint pain may develop and limit mobility for some individuals with SMCD. Lumbar lordosis may be present with typical onset by age three to five years.
Radiographic findings. The metaphyses of the long bones become flared or widened. The proximal and distal femoral and proximal tibial metaphyses are consistently affected (ragged, cupped, sclerotic, or splayed) with widening of the growth plates. Coxa vara (reduced angle to <120° between the head and the shaft of the femur) is seen in a majority of individuals and distinguishes SMCD from other forms of metaphyseal chondrodysplasia. Tibial and femoral bowing is typical. Metaphyseal irregularities of the distal ulnae and radii and enlarged capital femoral epiphyses are less commonly reported [Lachman et al 1988, Al Kaissi et al 2018]. The anterior rib ends are often cupped or sclerotic [Bateman et al 2005].
Hand involvement is present in fewer than half of individuals and is usually mild. Metaphyseal cupping of the distal metacarpals and proximal phalanges and shortening of the phalanges may be seen and are more pronounced in the fifth ray. Hand features may become less apparent with age [Elliott et al 2005].
Vertebral involvement is less common and, when present, is usually mild. Reported findings include platyspondyly and vertebral endplate anomalies (e.g., rounding of the anterior aspects of the vertebral bodies, superior and inferior indentations of the vertebral bodies, posterior wedging of the vertebrae) [Hasegawa et al 1994, Savarirayan et al 2000]. Vertebral changes may become less apparent with age.
Obesity. Limited mobility due to chronic joint pain may contribute to the development of obesity and associated comorbidities.
Craniofacial. The craniofacial bones and facial appearance are normal [Bateman et al 2005].
Neurodevelopment. Intelligence is normal. Attainment of early motor milestones is usually preserved in infancy; however, mild gross and fine motor delays may accompany orthopedic complications in early childhood.
Homozygotes. Biallelic pathogenic variants in COL10A1 have been associated with a more severe phenotype [Ain et al 2018] and in some cases embryonic lethality [Zhang et al 2018].
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Penetrance
Penetrance approaches 100%; however, there is wide inter- and intrafamilial phenotypic variation. Males and females are equally represented in the case literature. Normal stature has been reported, but radiographic features are usually still present. Age-dependent phenotypic manifestations are suggested by apparent reduction in hand and spine features with age [Savarirayan et al 2000, Elliott et al 2005].
Nomenclature
Spondylometaphyseal dysplasia (SMD) Japanese type. Affected individuals from one family reported had bowed legs, short stature, coxa vara, metaphyseal changes, and mild platyspondyly [Hasegawa et al 1994]. A heterozygous COL10A1 pathogenic variant was identified in affected individuals [Ikegawa et al 1998]. Expansion of the clinical phenotype of SMCD to include spinal changes led to the conclusion that SMD Japanese type and SMCD are the same disorder [Savarirayan et al 2000].
Prevalence
The exact prevalence of SMCD is unknown. Incidence has historically been estimated at between three and six cases per million [Gokhale & Mehta 2005]; however, these figures may be an underestimate [Al Kaissi et al 2018]. More than 130 unrelated individuals with SMCD and a confirmed pathogenic variant in COL10A1 have been reported.
Genetically Related (Allelic) Disorders
No phenotypes other than SMCD are known to be associated with germline pathogenic variants in COL10A1.
Differential Diagnosis
Table 3.
Other Genes of Interest in the Differential Diagnosis of Schmid Metaphyseal Chondrodysplasia
Rickets. Many individuals with SMCD receive an initial diagnosis of vitamin D-deficient rickets, due to a similar age of onset and overlapping clinical features, including genu varum or valgum, waddling gait, and nonspecific metaphyseal and epiphyseal irregularities. In rickets, coxa vara is usually absent and the distal femoral metaphyses are typically more affected. SMCD is distinguished from vitamin D-deficient rickets and metabolic bone disease on the basis of nutritional history and biochemical investigations (see Table 4).
Table 4.
Biochemical Findings in Schmid Metaphyseal Chondrodysplasia Compared with Vitamin D-Deficient Rickets and Metabolic Bone Diseases
Management
There are no published treatment or surveillance guidelines for Schmid metaphyseal chondrodysplasia (SMCD). Management should emphasize multidisciplinary care and a considered approach to surgical intervention when appropriate.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with SMCD, the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with Schmid Metaphyseal Chondrodysplasia
Treatment of Manifestations
Table 6.
Treatment of Manifestations in Individuals with Schmid Metaphyseal Chondrodysplasia
Surveillance
Table 7.
Recommended Surveillance for Individuals with Schmid Metaphyseal Chondrodysplasia
Agents/Circumstances to Avoid
Individuals with SMCD should maintain an appropriate weight for height, as obesity increases stress on the joints and may exacerbate joint pain and worsen the impact of genu varum and waddling gait on mobility. Education should include advice regarding weight loss (when appropriate), maintenance of a healthy diet, and regular low-impact exercise.
High-impact exercise, or exercise that causes repetitive strain on affected joints, should be avoided in favor of joint-friendly low-impact activities, including swimming and biking.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Carbamazepine is an FDA-approved medication for use in epilepsy and bipolar affective disorder. Its additional action as a stimulator of autophagy and proteasomal degradation pathways have led to its repurposing as a candidate therapy for conditions caused by retention of misfolded mutated structural proteins, such as SMCD [Hidvegi et al 2010]. In preclinical studies, carbamazepine alleviates endoplasmic reticulum stress (unfolded protein response) caused by the presence of structurally abnormal and misfolded collagen X in prehypertrophic chondrocytes, restoring growth and improving coxa vara in a validated mouse model of SMCD [Hidvegi et al 2010]. Carbamazepine received orphan drug designation by the European Commission for the treatment of SMCD in September 2016 (EMA/OD/148/16 and EMA/COMP/513538/2016).
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Schmid metaphyseal chondrodysplasia (SMCD) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Most individuals diagnosed with SMCD represent simplex cases (i.e., a single occurrence in a family) and have the disorder as the result of a de novo COL10A1 pathogenic variant. The proportion of cases caused by a de novo pathogenic variant is unknown.
- Some individuals diagnosed with SMCD have an affected parent [Ikegawa et al 1998, Hasegawa et al 2015, Zhang et al 2018]. The heterozygous parent almost always exhibits features of the condition; however, considerable intrafamilial phenotypic variability is observed in SMCD.
- Physical examination and measurement of proportions is recommended for the parents of a proband with an apparent de novo pathogenic variant. Recommendations may also include radiographs (as isolated asymptomatic individuals have been reported with radiographic changes only) and molecular genetic testing.
- If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent. Though theoretically possible, no instances of a proband inheriting a pathogenic variant from a parent with germline mosaicism have been reported.
- The family history of some individuals diagnosed with SMCD may appear to be negative because of failure to recognize the disorder in family members, reduced penetrance (reported in rare, isolated cases), or mild radiographic features in an affected parent that have not previously come to medical attention. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has been performed on the parents of the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs is 50%. Because considerable intrafamilial phenotypic variability is observed in SMCD, a heterozygous sib may have milder or more severe manifestations of SMCD.
- If the proband has a known SMCD-causing pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the COL10A1 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for SMCD because of the possibility of reduced penetrance in a heterozygous parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband
- Each child of an individual with SMCD has a 50% chance of inheriting the COL10A1 pathogenic variant.
- Because many individuals with short stature have reproductive partners with short stature, offspring of individuals with SMCD may be at risk of having double heterozygosity for two dominantly inherited bone growth disorders. The phenotypes of these individuals are distinct from those of the parents, and the affected individuals have serious sequelae and poor outcomes [Flynn & Pauli 2003].
- Biallelic COL10A1 pathogenic variants. A single individual with biallelic SMCD-causing pathogenic variants has been reported, with a more severe presentation, born to parents who were only mildly affected. Multiple miscarriages have been reported in a large consanguineous family segregating SMCD, leading to the proposal that homozygous COL10A1 pathogenic variants may result in early fetal demise.
- If the proband and the proband's reproductive partner are affected with different dominantly inherited skeletal dysplasias, each child has a 25% likelihood of average stature, a 25% likelihood of having the same skeletal dysplasia as the father, a 25% likelihood of having the same skeletal dysplasia as the mother, and a 25% likelihood of inheriting a pathogenic variant from both parents and being at risk for a poor outcome, including loss of pregnancy.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the COL10A1 pathogenic variant, his or her family members may be at risk.
Related Genetic Counseling Issues
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the pathogenic variant identified in the proband or clinical evidence of the disorder, the pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of affected individuals.
Prenatal Testing and Preimplantation Genetic Testing
Once the COL10A1 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Genetic and Rare Diseases Information Center (GARD)
- Little People UK
- Short Statured People of Australasia
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Schmid Metaphyseal Chondrodysplasia: Genes and Databases
Table B.
OMIM Entries for Schmid Metaphyseal Chondrodysplasia (View All in OMIM)
Molecular Pathogenesis
Type X collagen, encoded by COL10A1, is a homotrimer non-fibrillar collagen protein consisting of three α-1(X) chains. Type X collagen is present in the extracellular matrix and expressed exclusively by hypertrophic chondrocytes in the cartilage growth plates of growing bones undergoing endochondral ossification.
Pathogenic variants in COL10A1 are clustered in the C-terminal non-collagenous (NC1) domain, which contains motifs required for normal assembly of the collagen trimer. Both missense and truncating (frameshift and nonsense) variants in COL10A1 cause collagen X protein misfolding during protein synthesis, resulting in a failure of trimerization and aggregation within the endoplasmic reticulum (ER) of hypertrophic chondrocytes. Resultant ER stress, activation of the unfolded protein response, and reduced levels of functional type X collagen in the growth plate cause chondrodysplasia and development of the SMCD phenotype [Rajpar et al 2009].
Mechanism of disease causation. Both haploinsufficiency [Bateman et al 2003] and dominant-negative [Chan et al 1998, Wilson et al 2005] mechanisms have been previously proposed. Recognition of the central role of the unfolded protein response in molecular pathogenesis suggests a neomorphic, dominant-negative model in which the misfolding and accumulation of mutated type X collagen is responsible for this phenotype.
References
Literature Cited
- Ain NU, Mäkitie O, Naz S. Autosomal recessive chondrodysplasia with severe short stature caused by a biallelic COL10A1 variant. J Med Genet. 2018;55:403–7. [PubMed: 28830906]
- Al Kaissi A, Ghachem MB, Nabil NM, Kenis V, Melchenko E, Morenko E, Grill F, Ganger R, Kircher SG. Schmid's type of metaphyseal chondrodysplasia: diagnosis and management. Orthop Surg. 2018;10:241–6. [PMC free article: PMC6594483] [PubMed: 30027601]
- Bateman JF, Freddi S, Nattrass G, Savarirayan R. Tissue-specific RNA surveillance? Nonsense-mediated mRNA decay causes collagen X haploinsufficiency in Schmid metaphyseal chondrodysplasia cartilage. Hum Mol Genet. 2003;12:217–25. [PubMed: 12554676]
- Bateman JF, Wilson R, Freddi S, Lamandé SR, Savarirayan R. Mutations of COL10A1 in Schmid metaphyseal chondrodysplasia. Hum Mutat. 2005;25:525–34. [PubMed: 15880705]
- Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167A:2869–92. [PubMed: 26394607]
- Chan D, Weng YM, Graham HK, Sillence DO, Bateman JF. A nonsense mutation in the carboxyl-terminal domain of type X collagen causes haploinsufficiency in chmid metaphyseal chondrodysplasia. J Clin Invest. 1998;101:1490–9. [PMC free article: PMC508727] [PubMed: 9525992]
- Elliott AM, Field FM, Rimoin DL, Lachman RS. Hand involvement in Schmid metaphyseal chondrodysplasia. Am J Med Genet A. 2005;132A:191–3. [PubMed: 15578582]
- Flynn MA, Pauli RM. Double heterozygosity in bone growth disorders: four new observations and review. Am J Med Genet A. 2003;121A:193–208. [PubMed: 12923858]
- Gokhale S, Mehta S. Schmid type metaphyseal chondrodysplasia. Indian Pediatr. 2005;42:1252. [PubMed: 16424569]
- Hasegawa K, Higuchi Y, Yamashita M, Tanaka H. Japanese familial case with metaphyseal dysplasia, Schmid Type caused by the p.T555P mutation in the COL10A1 gene. Clin Pediatr Endocrinol. 2015;24:33–6. [PMC free article: PMC4322291] [PubMed: 25678758]
- Hasegawa T, Kozlowski K, Nishimura G, Hara H, Hasegawa Y, Aso T, Koto S, Nagai T, Tsuchiya Y. Japanese type of spondylo-metaphyseal dysplasia. Pediatr Radiol. 1994;24:194–7. [PubMed: 7936797]
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, Perlmutter DH. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–32. [PubMed: 20522742]
- Ikegawa S, Nishimura G, Nagai T, Hasegawa T, Ohashi H, Nakamura Y. Mutation of the type X collagen gene (COL10A1) causes spondylometaphyseal dysplasia. Am J Hum Genet. 1998;63:1659–62. [PMC free article: PMC1377637] [PubMed: 9837818]
- Lachman RS, Rimoin DL, Spranger J. Metaphyseal chondrodysplasia, Schmid type. Clinical and radiographic delineation with a review of the literature. Pediatr Radiol. 1988;18:93–102. [PubMed: 3281118]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5. [PMC free article: PMC3965032] [PubMed: 24234437]
- Mäkitie O, Susic M, Ward L, Barclay C, Glorieux FH, Cole WG. Schmid type of metaphyseal chondrodysplasia and COL10A1 mutations--findings in 10 patients. Am J Med Genet A. 2005;137A:241–8. [PubMed: 16088909]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Rajpar MH, McDermott B, Kung L, Eardley R, Knowles L, Heeran M, Thornton DJ, Wilson R, Bateman JF, Poulsom R, Arvan P, Kadler KE, Briggs MD, Boot-Handford RP. Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PLoS Genet. 2009;5:e1000691. [PMC free article: PMC2757901] [PubMed: 19834559]
- Savarirayan R, Cormier-Daire V, Lachman RS, Rimoin DL. Schmid type metaphyseal chondrodysplasia: a spondylometaphyseal dysplasia identical to the "Japanese" type. Pediatr Radiol. 2000;30:460–3. [PubMed: 10929364]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–77. [PMC free article: PMC5429360] [PubMed: 28349240]
- Wilson R, Freddi S, Chan D, Cheah KS, Bateman JF. Misfolding of collagen X chains harboring Schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation, intracellular retention, and activation of the unfolded protein response. J Biol Chem. 2005;280:15544–52. [PubMed: 15695517]
- Zhang C, Liu J, Iqbal F, Lu Y, Mustafa S, Bukhari F, Lou H, Fu R, Wu Z, Yang X, Bukhari I, Aslam M, Xu S. A missense point mutation in COL10A1 identified with whole-genome deep sequencing in a 7-generation Pakistan dwarf family. Heredity (Edinb). 2018;120:83–9. [PMC free article: PMC5837121] [PubMed: 29234170]
Chapter Notes
Acknowledgments
The authors wish to acknowledge the patients, their families, and the clinicians of the multidisciplinary bone dysplasia clinic at the Victorian Clinical Genetics Services and Royal Children's Hospital.
Revision History
- 21 October 2019 (sw) Review posted live
- 5 July 2019 (rs) Original submission
Publication Details
Author Information and Affiliations
Murdoch Children's Research Institute
Parkville, Victoria, Australia
Murdoch Children's Research Institute
Parkville, Victoria, Australia
Publication History
Initial Posting: October 21, 2019.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Richmond CM, Savarirayan R. Schmid Metaphyseal Chondrodysplasia. 2019 Oct 21. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
