Summary
Clinical characteristics.
TRIO-related neurodevelopmental disorder (TRIO-NDD) is characterized by two phenotypes: TRIO-NDD due to gain-of-function variants and TRIO-NDD due to loss-of-function variants.
TRIO-NDD due to gain-of-function variants within the spectrin repeat domain is characterized by moderate-to-severe developmental delay, intellectual disability, macrocephaly (or relative macrocephaly), neurobehavioral manifestations (poor attention, stereotypies, obsessive-compulsive behavior, aggressive behavior, and autism spectrum disorder), and early feeding difficulties with poor weight gain and growth deficiency. Seizures, constipation, scoliosis, dental abnormalities, and cardiac anomalies are also reported.
TRIO-NDD due to loss-of-function variants is characterized by mild-to-moderate developmental delay and intellectual disability, microcephaly, neurobehavioral manifestations (poor attention, aggressive behavior, autism spectrum disorder, obsessive-compulsive traits, and stereotypies), early feeding difficulties with poor weight gain, dental abnormalities, and digit anomalies, including 2-3 toe syndactyly in some individuals. Seizures, constipation, scoliosis, and cardiac anomalies are also reported.
Diagnosis/testing.
The diagnosis of TRIO-NDD is established in a proband with a heterozygous TRIO pathogenic variant identified by molecular genetic testing.
Management.
Treatment of manifestations: Treatment is symptomatic and includes routine management of developmental delays, intellectual disability, neurobehavioral manifestations, seizures, feeding difficulties, gastroesophageal reflux, constipation, spine abnormalities, and dental abnormalities, as well as for rarely occurring cardiovascular anomalies and recurrent infections.
Surveillance: At each visit, monitor developmental progress and educational needs; behavioral assessments for attention, aggression, and/or social communication difficulties; growth and feeding assessments to ensure optimal nutritional status; assessment for seizures, constipation, spine deformities, and frequent infections; regular dental evaluations.
Genetic counseling.
TRIO-NDD is an autosomal dominant disorder. The majority of individuals diagnosed with TRIO-NDD have the disorder as a result of a de novo pathogenic variant; approximately 15% inherited the TRIO pathogenic variant from an affected parent. TRIO gain-of-function missense variants (affecting the spectrin repeat domain) and TRIO loss-of-function missense variants (within the GEFD1 domain) are typically de novo. TRIO loss-of-function truncating variants may occur de novo or be inherited from an affected parent. Each child of an individual with TRIO-NDD has a 50% chance of inheriting the TRIO pathogenic variant. Once the TRIO pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
TRIO-related neurodevelopmental disorder (TRIO-NDD) should be considered in individuals with any combination of the following clinical findings:
- Early feeding issues with poor weight gain
- Delayed motor and speech development
- Variable intellectual functioning ranging from borderline cognitive function (IQ: 70-85) to severe intellectual disability
- Neurobehavioral manifestations including stereotypies, obsessive-compulsive behavior, autistic findings or autism spectrum disorder, attention-deficit/hyperactivity disorder, aggression, and/or sleep disorder
- Abnormal head circumference, either macrocephaly or microcephaly
- Minor digit anomalies including short and/or tapering fingers, broad proximal interphalangeal joints, clinodactyly of the fifth finger, and/or 2-3 toe syndactyly
- Dental anomalies, including dental crowding and delayed or failed tooth eruption
- Scoliosis and/or kyphosis
Establishing the Diagnosis
The diagnosis of TRIO-NDD is established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in TRIO identified by molecular genetic testing (see Table 1).
Note: (1) As per ACMG/AMP variant interpretation guidelines , the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic, and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variant" in this section is understood to include any likely pathogenic variant. (2) Identification of a heterozygous TRIO variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing in a child with developmental delay or an older individual with intellectual disability may begin with exome sequencing. Other options include genome sequencing or use of a multigene panel. Note: Single-gene testing (sequence analysis of TRIO, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended.
Molecular genetic testing approaches can include a combination of comprehensive genomic testing (exome sequencing, genome sequencing) and gene-targeted testing (multigene panel).
- Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is most commonly used and yields results similar to an intellectual disability multigene panel, with the additional advantage that exome sequencing includes genes recently identified as causing intellectual disability whereas some multigene panels may not. Genome sequencing is also possible.
- An intellectual disability multigene panel that includes TRIO and other genes of interest (see Differential Diagnosis) may also be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Of note, given the rarity of TRIO-NDD, some panels for intellectual disability may not include this gene. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 1.
Molecular Genetic Testing Used in TRIO-Related Neurodevelopmental Disorder
Clinical Characteristics
Clinical Description
TRIO-related neurodevelopmental disorder (TRIO-NDD) is characterized by two phenotypes with some overlapping clinical features: TRIO-NDD due to gain-of-function variants and TRIO-NDD due to loss-of-function variants. Individuals with gain-of-function variants often have more severe developmental delay and intellectual disability as well as macrocephaly. Individuals with TRIO loss-of-function variants have mild-to-moderate developmental delay and often have microcephaly. (See Table 2 for other common and distinguishing features.) To date, 56 individuals have been identified with a pathogenic variant in TRIO and for whom sufficient clinical information was available for the current review [Ba et al 2016; Pengelly et al 2016; Barbosa et al 2020; Schultz-Rogers et al 2020; Kloth et al 2021; Gazdagh et al, unpublished data].
Table 2.
Select Features of TRIO-Related Neurodevelopmental Disorder
TRIO-NDD due to Gain-of-Function Variants
Developmental delay. The majority of individuals with TRIO gain-of-function variants are reported to have moderate-to-severe developmental delay affecting motor, speech, and cognition.
Among all individuals for whom information on independent sitting was available, approximately one third (6/17) achieved sitting by age 14 months. Seventy-five percent (13/17) achieved sitting by 36 months. For one affected individual, independent sitting occurred after age six years [Kloth et al 2021]. Two individuals, age 24 months and age three years one month, respectively, were not yet able to sit independently [Barbosa et al 2020, Kloth et al 2021]. Upon last assessment, walking was attained for seven individuals, with onset of walking ranging from 20 months to seven years [Barbosa et al 2020; Gazdagh et al, unpublished data]. Of those who walked independently, 70% did so by age three years and 80% by age five years. Among the ten individuals who were nonambulatory at last assessment, the oldest were age six to ten years [Barbosa et al 2020; Kloth et al 2021; Gazdagh et al, unpublished data].
Information on use of first words was available for 16 individuals, seven of whom were verbal. Earliest use of first words was at age 12 to 15 months [Gazdagh et al, unpublished data], and oldest age at first words was 5.5 years [Barbosa et al 2020]. Individuals who were nonverbal were ages 24 months to ten years two months [Gazdagh et al, unpublished data].
Intellectual disability is likely present in all individuals. However, no formal IQ assessments have been reported in individuals with TRIO gain-of-function variants.
Neurobehavioral manifestations. Among individuals with a TRIO gain-of-function variant, autism spectrum disorder or autistic findings were reported in 27% (4/15 individuals), including three individuals who had a formal diagnosis of autism spectrum disorder. Poor attention was reported in 56% (9/16), stereotypies in 47% (8/17), and obsessive-compulsive behavior in 47% (7/15). Aggressive behavior was a feature in 29% (5/17), with one of these individuals described as having temper tantrums. Disrupted sleep has been described in a few individuals in the literature although it is likely underreported [Varvagiannis, personal communication].
Seizures occurred in 37% of individuals (7/19) for whom information was available. Variable types of seizures have been reported, including nocturnal seizures in two individuals, absence seizures, and myoclonic movements of the extremities and eyelids [Barbosa et al 2020; Kloth et al 2021; Gazdagh et al, unpublished data]. One of these individuals developed seizures at age 30 years [Barbosa et al 2020]. Two further individuals had abnormal EEGs or suspected drop seizures not captured on EEG recordings [Barbosa et al 2020]; thus, seizures may be present in up to 47% (9/17 individuals).
Macrocephaly. TRIO gain-of-function variants are often associated with absolute macrocephaly (61% of individuals) [Barbosa et al 2020; Kloth et al 2021; Gazdagh et al, unpublished data]. Occipital frontal circumference (OFC) three or more standard deviations (SD) above the mean has been reported in four individuals. One of these individuals had an OFC of 4.7 SD above the mean [Barbosa et al 2020]. An additional two individuals were reported to have relative macrocephaly [Kloth et al 2021; Gazdagh et al, unpublished data].
Additional neurologic manifestations included tremor in two individuals, dystonia in two individuals, and ataxia and/or wide-based ataxic gait in two individuals [Barbosa et al 2020; Gazdagh et al, unpublished data]. One individual had an EMG suggestive of very mild demyelinating peripheral neuropathy [Barbosa et al 2020].
Brain MRI abnormalities. Although the total number of individuals who have had a brain MRI is not known, abnormal brain imaging was reported in five individuals. Four individuals had dilated ventricles [Barbosa et al 2020; Kloth et al 2021; Gazdagh et al, unpublished data]. Two were described as having enlarged inner and outer cerebrospinal fluid spaces, and two were diagnosed with hydrocephalus (one of these individuals had a history of intraventricular hemorrhage in the neonatal period) [Gazdagh et al, unpublished data]. Abnormal corpus callosum was a feature in three individuals, in two of whom the corpus callosum was thin [Barbosa et al 2020; Gazdagh et al, unpublished data], and in one there was hypoplasia/agenesis of the corpus callosum [Kloth et al 2021]. Delayed myelin maturation, Arnold-Chiari malformation, loss of white matter volume, and dysgenesis of anterior commissure with colpocephaly were among variable findings within these five individuals.
Feeding difficulties. Neonatal and infantile feeding difficulties including poor suck, impaired bottle feeding, swallowing difficulties, and frequent vomiting or regurgitation are common, resulting in poor weight gain. Silent aspiration was reported in one individual. Gastrointestinal reflux appears to contribute to these difficulties in some individuals. Feeding difficulties required nutritional support in approximately 30% of individuals, routinely by tube feeding (gastrostomy or less frequently nasogastric). Poor weight gain and early growth deficiency was reported in 80% of infants for whom this information was available.
Growth. The majority of older individuals continued to have poor weight gain; in 67% of individuals (12/18) weight on last examination was reported to be two or more SD below the mean; no individuals with gain-of-function variants were reported to have weight two or more SD above the mean. Short stature (height ≥2 SD below the mean) was reported in 31% of individuals (5/16).
Gastrointestinal manifestations. Gastroesophageal reflux may contribute to feeding difficulties, although the exact frequency is not known. Episodic vomiting has been reported in a few individuals [Barbosa et al 2020]. Constipation appears to be frequent, occurring in 38% of reported individuals. Rare findings reported in single individuals include intestinal malrotation requiring surgery as well as eosinophilic esophagitis [Gazdagh et al, unpublished data].
Musculoskeletal features. Scoliosis was reported in 44% of individuals [Barbosa et al 2020; Kloth et al 2021; Gazdagh et al, unpublished data]. In two individuals, abnormal spine curvature occurred early and/or necessitated surgery [Barbosa et al 2020, Kloth et al 2021]. Kyphosis was described in two individuals. Short and/or tapering fingers were described in 35% of individuals.
Dysmorphic facial features. Barbosa et al [2020] analyzed facial photos of affected individuals using quantitative facial phenotyping. Comparisons of facial features were performed between all individuals with TRIO-NDD and controls and between those with TRIO gain-of-function or loss-of-function variants and controls. Significant clustering was only observed between persons with a TRIO gain-of-function variant compared to matched controls, leading to the suggestion of a distinctive facial gestalt.
Presence of a tall forehead and prominent ears appear to be common [Gazdagh, personal communication]. In the cohort of 20 individuals, the following features were described in more than one individual:
- Prominent or tall forehead in six individuals, with frontal bossing in six additional individuals
- Highly arched eyebrows reported in three individuals. with synophrys reported in two
- Hypertelorism in two individuals, and downslanted palpebral fissures in two individuals
- Low-set ears in three individuals, and large ear lobes in two individuals
- Wide mouth, everted vermilion of the upper lip, and high palate in two individuals each
- Micro-/retrognathia in two individuals
Dental abnormalities. Among the 15 individuals evaluated for dental anomalies, five had relevant findings. These included advanced eruption in one individual [Barbosa et al 2020] and delayed eruption in one individual [Gazdagh et al, unpublished data]. One additional individual had absence of the second molars at age 37 months [Gazdagh et al, unpublished data]. Dental crowding was present in three individuals [Barbosa et al 2020; Gazdagh et al, unpublished data].
Cardiac anomalies were present in 11% of individuals. These included one individual with bicuspid aortic valve, aortic regurgitation, and prominent aortic root [Gazdagh et al, unpublished data], and a second individual with atrioventricular septal defect, patent ductus arteriosus, and type A interrupted aortic arch type.
Genitourinary anomalies include three individuals with neurogenic bladder [Gazdagh et al, unpublished data] and two individuals with enuresis, one of whom had recurrent urinary tract infections.
Other
- Supernumerary or inverted nipples (each reported in a single individual) [Gazdagh et al, unpublished data]
- Multiple hyperpigmented nevi (one individual) [Kloth et al 2021]
- Frequent airway infections (one individual) [Kloth et al 2021]
TRIO-NDD due to Loss-of-Function Variants
Developmental delay. Delayed attainment of milestones is reported in all individuals. Mild-to-moderate developmental delay is typically observed. Severe delay appears to be very rare.
Independent sitting was achieved in 19 individuals, in 80% of these by the age of 14 months. Two individuals were not sitting independently at their last assessment (ages 21 months and 16 years, respectively) [Barbosa et al 2020; Gazdagh et al, unpublished data]. The majority of individuals were also ambulatory (20/24). The range for onset of walking was age 12 to 36 months [Barbosa et al 2020], with 80% of individuals ambulatory by age 22 months. Four individuals were nonambulatory at last assessment (ages 20 months, 21 months, 3.5 years, and 16 years, respectively) [Barbosa et al 2020; Gazdagh et al, unpublished data].
First words occurred by the age of 48 months in 80% of the 20 individuals who were verbal. Four individuals were nonverbal at last assessment (ages 21 months, 3.5 years, 14 years, and 16 years, respectively) [Barbosa et al 2020; Kolbjer et al 2021; Gazdagh et al, unpublished data]. The individual who was nonverbal at age 16 years had an intragenic in-frame multiexon TRIO deletion spanning the GEFD1 domain [Kolbjer et al 2021; Gazdagh et al, unpublished data].
Intellectual disability. Most individuals have intellectual disability. Formal IQ assessment has been reported in only four individuals. Two individuals had borderline intellectual functioning (IQ scores of 81 and 78, respectively); two individuals had mild intellectual disability (IQ scores of 62 and 68, respectively) [Ba et al 2016].
Behavioral phenotype. Behavioral findings have been reported in 94% of individuals. Poor attention was reported in 79% (22/28); at least three individuals were diagnosed with attention-deficit/hyperactivity disorder. Obsessive-compulsive findings were reported in 45% of individuals (8/23). Aggressive behavior was reported in 44% (12/27); in two of these individuals, self-mutilating behaviors were reported [Ba et al 2016; Gazdagh et al, unpublished data]. Autism spectrum disorder or autistic findings were described in 40% (10/27); four of these individuals received a formal diagnosis of autism spectrum disorder or pervasive developmental disorder not otherwise specified. Approximately 25% (6/26) presented with stereotypies. In one individual this manifested as echolalia and "balancing" (no additional description of "balancing" was included in the report) [Barbosa et al 2020]. Hand flapping was a feature in an additional individual [Schultz-Rogers et al 2020]. Disrupted sleep has been described in a few individuals in the literature, although it is likely underreported [Varvagiannis, personal communication].
Seizures were reported in 24% of individuals with a TRIO loss-of-function variant. Seizures were more common in individuals with a truncating variant (30%-35%). Only one in ten individuals with a TRIO missense variant in the GEFD1 domain (see Molecular Genetics) had seizures. Reported seizure types included febrile seizures during early childhood, myoclonic seizures, and absence seizures with eyelid and forehead myoclonus refractory to treatment [Schultz-Rogers et al 2020; Gazdagh et al, unpublished data]. One individual had various types of seizures at age four years [Barbosa et al 2020]. An adolescent with a TRIO intragenic deletion had severe developmental delay and a history of refractory seizures.
Microcephaly. Approximately 70% of individuals (25/35) had microcephaly [Ba et al 2016, Pengelly et al 2016, Barbosa et al 2020, Schultz-Rogers et al 2020]. Severe microcephaly (>3 SD below the mean) was reported in at least 15 individuals, including three individuals with an OFC average of 6 SD below the mean [Barbosa et al 2020; Gazdagh et al, unpublished data].
Note: Macrocephaly was reported in two individuals with a TRIO loss-of function variant [Schultz-Rogers et al 2020; Gazdagh et al, unpublished data].
Brain MRI abnormalities. Although the total number of individuals who have had a brain MRI is not known, six individuals had abnormal brain imaging. In three individuals there was partial agenesis or hypoplasia of the corpus callosum [Barbosa et al 2020; Gazdagh et al, unpublished data]. Ventricular anomalies were found in two individuals, including abnormal aspect of the lateral ventricle and ventriculomegaly [Gazdagh et al, unpublished data]. Other anomalies observed in single individuals included progressive leukoencephalopathy, lissencephaly, and interhemispheric cyst [Barbosa et al 2020; Gazdagh et al, unpublished data].
Feeding difficulties. Neonatal and infantile feeding difficulties including poor suck, impaired bottle feeding, and swallowing difficulties are common and result in poor weight gain. Gastrointestinal reflux appears to contribute to these difficulties in some individuals. Feeding difficulties required nutritional support (e.g., tube feeding) in 22% of individuals. Poor weight gain and early growth deficiency was reported in 72% of infants for whom this information was available.
Growth. Some individuals had persistent poor weight gain; in 30% of individuals (8/27), weight on last examination was reported to be two or more SD below the mean. However, 11% individuals (3/27) with loss-of-function variants were reported to have weight two or more SD above the mean. Short stature (height ≥2 SD below the mean) was reported in 14% of individuals (4/29).
Gastrointestinal manifestations. Gastroesophageal reflux may contribute to feeding difficulties, although the exact frequency is not known. Episodic vomiting has been reported in a few individuals [Barbosa et al 2020]. Constipation may be more frequent in individuals with a TRIO loss-of-function missense variant (7/11; 63%) compared to those with a truncating variant (5/14; 36%). One individual with a TRIO intragenic deletion had an anal fistula [Ba et al 2016].
Toe syndactyly. Five individuals were reported to have 2-3 toe syndactyly [Barbosa et al 2020; Gazdagh et al, unpublished data].
Short and/or tapering fingers are described in 45% of individuals (14/31).
Additional digit anomalies. Less common digit anomalies reported in individuals with TRIO loss-of-function variants included swelling (or broad) proximal interphalangeal joints (five individuals), fifth finger clinodactyly (five individuals), and proximally placed thumb (one individual).
Other musculoskeletal features. Scoliosis was less frequent in individuals with loss-of-function variants and was observed in 15% (4/27). Kyphosis was described in two individuals. Pectus excavatum was reported in two individuals [Ba et al 2016; Gazdagh et al, unpublished data].
Dysmorphic facial features. Barbosa et al [2020] analyzed facial photos of affected individuals using quantitative facial phenotyping. Comparisons of facial features were performed between all individuals with TRIO-NDD and controls and between individuals with TRIO gain-of-function or loss-of-function variants and controls. Significant clustering was not identified in persons with loss-of-function variants.
Upslanted palpebral fissures, tubular nose, and bulbous nose appear to be common in individuals with a TRIO loss-of-function variant [Gazdagh, personal communication]. Among the 36 individuals reported, the following dysmorphic features were described: facial asymmetry (five individuals), tall forehead (three individuals), broad forehead (one individual), synophrys (four individuals), highly-arched eyebrows (one individual), thick or thick and straight eyebrows (two individuals), upslanted palpebral fissures (three individuals), hypertelorism (two individuals), large and/or protruding ears (seven individuals), abnormal ear helices (two individuals), abnormal nasal configuration (most individuals), thin vermilion of the upper lip (four individuals), thick vermilion of upper and lower lips (three individuals), high palate (five individuals), and micro-/retrognathia (at least eight individuals).
Dental anomalies. Approximately 60% of individuals had dental anomalies, including delayed eruption in 30% (7/24) and advanced eruption in one of 24 individuals. Dental crowding was present in 30% (8/27). Hypodontia/oligodontia was reported in two sibs. One additional individual had hypodontia, although this was also present in an unaffected sib [Barbosa et al 2020]. Other findings included abnormality of the enamel/caries (three individuals), macrodontia of incisors (two individuals), and microdontia (one individual).
Cardiovascular abnormalities. Structural defects were reported in 12% of affected individuals (3/26). These included two individuals with an atrial septal defect [Barbosa et al 2020; Gazdagh et al, unpublished data] and tetralogy of Fallot in one individual [Gazdagh et al, unpublished data]. Arrhythmia has been reported in two individuals with TRIO truncating variants. While in one individual presence of the conduction defect was suspected [Gazdagh et al, unpublished data]; the second individual had an additional KCNJ2 pathogenic variant that could be the cause of the arrhythmia [Pengelly et al 2016].
Ophthalmologic manifestations were reported in four individuals. Each of the following were reported in one individual each: visual problems suggestive of bilateral macular pathway dysfunction with pale optic discs, total optic atrophy and poor vision, strabismus, and nasolacrimal duct obstruction [Barbosa et al 2020; Gazdagh et al, unpublished data].
Other
- Recurrent infections (three individuals) [Ba et al 2016]
- Urinary incontinence (two individuals), although one had syringomyelia [Barbosa et al 2020; Gazdagh et al, unpublished data]
- Brisk reflexes (two individuals) [Ba et al 2016; Gazdagh et al, unpublished data]
- Fatigue (two individuals) [Ba et al 2016]
- Bilateral accessory nipples (one individual) [Gazdagh et al, unpublished data]
- Unilateral cleft lip (one individual) [Gazdagh et al, unpublished data]
- Profound neonatal anemia (one individual) [Gazdagh et al, unpublished data]
- Cutis aplasia (one individual) [Schultz-Rogers et al 2020]
Genotype-Phenotype Correlations
Gain-of-function missense variants (affecting the spectrin repeat domain) are associated with more severe developmental delay and intellectual disability, macrocephaly, and a higher risk of seizures and scoliosis than individuals with loss-of-function variants. Pathogenic variants involving p.Arg1078 were reported to be associated with the most severe phenotype [Barbosa et al 2020].
Loss-of-function missense variants within the GEFD1 domain as well as truncating variants throughout the gene cause less severe intellectual disability than gain-of-function variants and microcephaly in some individuals.
A summary of major developmental milestones in individuals with a TRIO gain-of-function variant (within the spectrin domain) or loss-of-function variant (within the GEFD1 domain or a truncating variant) is available here (pdf).
- Barbosa et al [2020] commented that truncating variants are associated with more variable phenotypes.
- The developmental phenotype of nonsense, frameshift, and splicing variants was often less severe compared to missense variants within the GEFD1 domain. A similar conclusion might be indirectly inferred by the fact that missense variants within the GEFD1 domain are de novo in all individuals for whom inheritance was available (18/18), while only 60% (10/17) of truncating variants are de novo.
- Loss-of-function truncating variants are associated with a higher risk of seizures (30%-35%) than missense variants in the GEFD1 domain (likely 10%).
- Delayed dental eruption and dental crowding are more common in individuals with missense variants within the GEFD1 domain and truncating variants [Barbosa et al 2020]. Advanced eruption was seen in one individual with a gain-of-function variant [Barbosa et al 2020] and another with a loss-of-function variant [Gazdagh et al, unpublished data].
- Toe syndactyly (2-3) was only reported in individuals with missense variants within the GEFD1 domain and truncating variants.
- Broad (or swelling of) proximal interphalangeal joints and fifth finger clinodactyly were only reported in individuals with missense variants within the GEFD1 domain and truncating variants.
Penetrance
Penetrance appears to be complete. Based on the seven individuals (from five families) in whom the TRIO pathogenic variant was inherited [Pengelly et al 2016; Ba et al 2016; Schultz-Rogers et al 2020; Gazdagh et al, unpublished data], the parent may be similarly [Ba et al 2016] or only mildly affected [Schultz-Rogers et al 2020] compared to the index case within a family.
Prevalence
TRIO-NDD appears to be rare and accounts for fewer than 1% of individuals with intellectual disability and/or autism spectrum disorder.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in TRIO.
Differential Diagnosis
Developmental delay, intellectual disability, and/or neurobehavioral manifestations and abnormal head circumference are among the major features in TRIO-related neurodevelopmental disorder (TRIO-NDD) for which affected individuals may be referred for genetic evaluation. Because these features are not sufficient to diagnose TRIO-NDD, all intellectual developmental disorders without other distinctive findings should be considered in the differential diagnosis. See OMIM Autosomal Dominant, Autosomal Recessive, Nonsyndromic X-Linked, and Syndromic X-Linked Intellectual Developmental Disorder Phenotypic Series.
Management
No clinical practice guidelines for TRIO-related neurodevelopmental disorder (TRIO-NDD) have been published.
Evaluations and Referrals Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with TRIO-NDD, the evaluations and referrals summarized in Table 3 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis of TRIO-Related Neurodevelopmental Disorder
Treatment of Manifestations
Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 4).
Table 4.
Treatment of Manifestations in Individuals with TRIO-Related Neurodevelopmental Disorder
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the US; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., scoliosis).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and is typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 5 are recommended.
Table 5.
Recommended Surveillance for Individuals with TRIO-Related Neurodevelopmental Disorder
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
TRIO-related neurodevelopmental disorder (TRIO-NDD) – encompassing TRIO-NDD due to gain-of-function variants and TRIO-NDD due to loss-of-function variants – is an autosomal dominant disorder.
- TRIO gain-of-function missense variants (affecting the spectrin repeat domain) and TRIO loss-of-function missense variants within the GEFD1 domain are typically de novo.
- TRIO loss-of-function truncating variants may occur de novo or be inherited from an affected parent.
Risk to Family Members
Parents of a proband
- The majority of individuals diagnosed with TRIO-NDD have the disorder as the result of a de novo pathogenic variant.
- Some individuals diagnosed with TRIO-NDD have an affected parent. Cumulative data on 56 individuals show that the pathogenic variant was inherited from an affected parent in roughly 15% of individuals [Ba et al 2016; Pengelly et al 2016; Barbosa et al 2020; Schultz-Rogers et al 2020; Kloth et al 2021; Gazdagh et al, unpublished data].
- Because TRIO-NDD is likely underdiagnosed, the actual proportion of simplex cases (defined as individuals with no obvious family history) and familial cases (defined as the presence of ≥2 related affected individuals) cannot be determined.
- Molecular genetic testing for the TRIO pathogenic variant identified in the proband is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
- The family history of some individuals diagnosed with TRIO-NDD may appear to be negative as a result of failure to recognize the disorder in family members. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has been performed on the parents of the proband.
Sibs of a proband. The risk to sibs of a proband depends on the genetic status of the proband's parents:
- If a parent of the proband has the TRIO pathogenic variant, the risk to the sibs of inheriting the variant is 50%.
- Clinical variability has been observed among affected family members with the same TRIO truncating variant [Ba et al 2016; Pengelly et al 2016; Schultz-Rogers et al 2020; Gazdagh et al, unpublished data] (see also Genotype-Phenotype Correlations).
- If the TRIO pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the empiric recurrence risk to sibs is approximately 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016]. Recurrence due to parental germline mosaicism has not been reported to date.
Offspring of a proband. Each child of an individual with TRIO-NDD has a 50% chance of inheriting the TRIO pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the TRIO pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the TRIO pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Team TRIO: Support network for families and professionals
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968
- CDC - Child DevelopmentPhone: 800-232-4636
- MedlinePlus
- VOR: Speaking out for people with intellectual and developmental disabilitiesPhone: 877-399-4867Email: info@vor.net
- Human Disease Gene Website Series - Registry
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
TRIO-Related Neurodevelopmental Disorder: Genes and Databases
Table B.
OMIM Entries for TRIO-Related Neurodevelopmental Disorder (View All in OMIM)
Molecular Pathogenesis
TRIO encodes the triple functional domain protein (TRIO) consisting of several domains, notably an N-terminal SEC14 domain, several spectrin repeats, two Dbl-homology-Pleckstrin-homology (DH-PH) Rho-guanine exchange factor (GEF) domains, and a C-terminal serine/threonine kinase domain. The first DH-PH domain activates Rac1 and RhoG, while the second acts on RhoA. The protein product of TRIO, through its two GEF domains, acts as a Rho GTPase regulator.
TRIO is a major regulator of different processes, including cytokinesis, cell migration, axon guidance, and dendritic arborization, and is involved in synaptogenesis by modulating excitatory synaptic transmission [Barbosa et al 2020]. This role is mediated by Rac1 activation and actin cytoskeleton remodeling.
Pathogenic missense variants occur in two hot spots: missense variants in the spectrin repeat domain increase Rac1 activation, and missense variants in the first GEF domain (GEFD1) decrease Rac1 activation (see Figure 1). The aberrant levels of Rac1 activation result in two distinct phenotypes caused by TRIO pathogenic variants [Barbosa et al 2020].
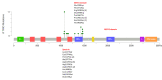
Mechanism of disease causation. Missense variants within the spectrin repeat domain result in gain of function (increased Rac1 activation).
Missense variants in the GEFD1 domain as well as truncating variants throughout the gene result in loss of function (decreased Rac1 activation).
TRIO-specific laboratory technical considerations. Gain-of-function variants reported to date localize within a range of residues (p.Leu1071 to Glu1159 [NM_007118.4; NP_009049.2]) of the broader spectrin repeat domain. Loss of function is the underlying mechanism for pathogenic missense variants within the GEFD1 domain, the latter spanning residues p.Arg1292 to p.Gln1591 (NM_007118.4; NP_009049.2).
Loss-of-function (truncating) variants have been observed in large-scale reference population data sets (e.g., the Exome Aggregation Consortium [ExAC] data set) [Lek et al 2016]. Few of these truncating variants are located in the last exon, presumably escaping nonsense-mediated decay [Ba et al 2016].
Chapter Notes
Author Notes
Dr Gazdagh (ku.shn.shu@hgadzag.alleirbag) and Prof Baralle (ku.ca.notos@ellarab.d) are actively involved in clinical research regarding individuals with TRIO-related neurodevelopmental disorder (TRIO-NDD). They would be happy to communicate with persons who have any questions regarding diagnosis of TRIO-NDD or other considerations.
Contact Dr Gazdagh and Dr Varvagiannis to inquire about review of TRIO variants of uncertain significance.
Acknowledgments
The authors are grateful to the patients and families who participated in these research projects, as well as the clinicians and scientists contributing to understanding of the clinical and molecular aspects of TRIO-NDD.
Revision History
- 23 March 2023 (sw) Comprehensive updated posted live
- 10 August 2017 (bp) Review posted live
- 23 December 2016 (kv) Original submission
References
Literature Cited
- Ba W, Yan Y, Reijnders MR, Schuurs-Hoeijmakers JH, Feenstra I, Bongers EM, Bosch DG, De Leeuw N, Pfundt R, Gilissen C, De Vries PF, Veltman JA, Hoischen A, Mefford HC, Eichler EE, Vissers LE, Nadif Kasri N, De Vries BB. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum Mol Genet. 2016;25:892–902. [PMC free article: PMC4754042] [PubMed: 26721934]
- Barbosa S, Greville-Heygate S, Bonnet M, Godwin A, Fagotto-Kaufmann C, Kajava AV, Laouteouet D, Mawby R, Wai HA, Dingemans AJM, Hehir-Kwa J, Willems M, Capri Y, Mehta SG, Cox H, Goudie D, Vansenne F, Turnpenny P, Vincent M, Cogné B, Lesca G, Hertecant J, Rodriguez D, Keren B, Burglen L, Gérard M, Putoux A. C4RCD Research Group; Cantagrel V, Siquier-Pernet K, Rio M, Banka S, Sarkar A, Steeves M, Parker M, Clement E, Moutton S, Tran Mau-Them F, Piton A, de Vries BBA, Guille M, Debant A, Schmidt S, Baralle D. Opposite modulation of RAC1 by mutations in TRIO is associated with distinct, domain-specific neurodevelopmental disorders. Am J Hum Genet. 2020;106:338–55. [PMC free article: PMC7058823] [PubMed: 32109419]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. [PMC free article: PMC3956037] [PubMed: 22588877]
- Kloth K, Graul-Neumann L, Hermann K, Johannsen J, Bierhals T, Kortüm F. More evidence on TRIO missense mutations in the spectrin repeat domain causing severe developmental delay and recognizable facial dysmorphism with macrocephaly. Neurogenetics. 2021;22:221–4. [PubMed: 34013494]
- Kolbjer S, Martin DA, Pettersson M, Dahlin M, Anderlid BM. Lissencephaly in an epilepsy cohort: Molecular, radiological and clinical aspects. Eur J Paediatr Neurol. 2021;30:71–81. [PubMed: 33453472]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. [PMC free article: PMC5018207] [PubMed: 27535533]
- Pengelly RJ, Greville-Heygate S, Schmidt S, Seaby EG, Jabalameli MR, Mehta SG, Parker MJ, Goudie D, Fagotto-Kaufmann C, Mercer C, Debant A, Ennis S, Baralle D, et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J Med Genet. 2016;53:735–42. [PMC free article: PMC5264232] [PubMed: 27418539]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Schultz-Rogers L, Muthusamy K, Pinto E, Vairo F, Klee EW, Lanpher B. Novel loss-of-function variants in TRIO are associated with neurodevelopmental disorder: case report. BMC Med Genet. 2020;21:219. [PMC free article: PMC7654171] [PubMed: 33167890]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
Publication Details
Author Information and Affiliations
Cyprus Institute of Neurology & Genetics
Cyprus, Greece
Radboud University Medical Center
Nijmegen, the Netherlands
Southampton General Hospital
Southampton, United Kingdom
Radboud University Medical Center
Nijmegen, the Netherlands
Southampton, United Kingdom
Publication History
Initial Posting: August 10, 2017; Last Update: March 23, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Varvagiannis K, Vissers LELM, Baralle D, et al. TRIO-Related Neurodevelopmental Disorder. 2017 Aug 10 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
.&p=BOOKS&id=447257_trio-id-Image001.jpg "Click on image to zoom")