

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.

Summary
Sequin is a stand-alone sequence record editor, designed for preparing new sequences for submission to GenBank and for editing existing records. Sequin runs on the most popular computer platforms found in biology laboratories, including PC, Macintosh, UNIX, and Linux. It can handle a wide range of sequence lengths and complexities, including entire chromosomes and large datasets from population or phylogenetic studies. Sequin is also used within NCBI by the GenBank and Reference Sequence indexers for routine processing of records before their release.
Sequin has a modular construction, which simplifies its use, design, and implementation. Sequin relies on many components of the NCBI Toolkit and thus acts as a quality assurance that these functions are working properly.
Detailed information on how to use Sequin to submit records to GenBank or edit sequence records can be found in the Sequin Quick Guide. Although this chapter will make frequent reference to that help document, the focus will be mostly on the underlying concepts and software components upon which Sequin is built.
Sequin: A Brief Overview
As input, Sequin takes a biological sequence(s) from a scientist wanting to submit or edit sequence data. The sequence (or set of sequences) can be new information that has not yet been assigned a GenBank Accession number, or it can be an existing GenBank sequence record. If Sequin is being used to submit a sequence(s) to GenBank, then the scientist is prompted to include his/her contact information, information about other authors, and the sequence, at the start of the submission process. Once all the necessary information has been entered, it is then possible to view the sequence in a variety of displays and edit it using Sequin's suite of editing tools.
Sequin is designed for use by people with different levels of expertise. Thus, it has several built-in functions that can, for example, ensure that a new user submits a valid sequence record to GenBank, or it can be prompted to automatically generate a sequence definition line. At the other end of the scale, for computer-literate users, Sequin can be customized by the addition of more (perhaps research-specific) analysis functions. Furthermore, there are some extremely powerful functions built into Sequin that are only available to NCBI Indexing staff. These are switched off by default in the public download version of Sequin because they include the ability to make the kinds of changes to a sequence record that can also completely destroy it, if handled incorrectly. These various built-in Sequin functions are discussed further below.
Sequin's versatility is based on its design: (a) Sequin holds the sequence(s) being manipulated in memory, in a structured format that allows a rapid response to the commands initiated by the person who is using Sequin; and (b) it makes use of many standard functions found in the NCBI Toolkit for both basic data manipulations and as components of Sequin-specific tasks. In particular, Sequin makes heavy use of the Toolkit's object manager, a “behind the scenes” support system that keeps track of Sequin's internal data structures and the relationships of each piece of information to others. This allows many of Sequin's functions to operate independently of each other, making data manipulation much faster and making the program easier to maintain.
Sequence Submission
Sequin is used to edit and submit sequences to GenBank and handles a wide range of sequence lengths and complexities. After downloading and installing Sequin, a scientist wanting to submit or edit a sequence(s) is led through a series of forms to input information about the sequence to be submitted. The forms are “smart”, and different forms will appear, customized to the type of submission. Detailed information on how to fill in these forms can be found within the Help feature of the Sequin application itself and in the online Help documents.
Sequin expects sequence data in FASTA formatted files, which should be prepared as plain text before uploading them into Sequin. Population, phylogenetic, and mutation studies can also be entered in PHYLIP, NEXUS, MACAW, or FASTA+GAP formats.
A sequence in FASTA format consists of a definition line, which starts with a “>”, and the sequence itself, which starts on a new line directly below the definition line. The definition line should contain the name or identifier of the sequence but may also include other useful information. In the case of nucleotides, the name of the source organism and strain should be included; for proteins, it is useful to include the gene and protein names. Given all this information, Sequin can automatically assemble a record suitable for inclusion in GenBank (see below). Detailed information on how to prepare FASTA files for Sequin can be found in the Quick Guide.
Single Sequences
For single nucleotide sequence submissions to GenBank, the submitter supplies Sequin with the nucleotide sequence and any translated protein sequence(s). For example, a submission consisting of a nucleotide from mouse strain BALB/c that contains the β-hemoglobin gene, encoding the adult major chain β-hemoglobin protein, would have two sequences with the following definition lines, where “BALB23g” and “BALB23p” are nucleotide and protein IDs provided by the submitter:
> BALB23g [organism=Mus musculus] [strain=BALB/c]
> BALB23p [gene=Hbb-b1] [protein=hemoglobin, beta adult major chain]
The organism name is essential to make a legal GenBank flatfile. It can be included in the definition line as shown above, for the convenience of the submitter, or one of the Sequin submission forms will prompt for its clarity.
Although it is not necessary to include a protein translation with the nucleotide submission, scientists are strongly encouraged to do so because this, along with the source organism information, enables Sequin to automatically calculate the coding region (CDS) on the nucleotide being submitted. Furthermore, with gene and protein names properly annotated, the record becomes informative to other scientists who may retrieve it through a BLAST or Entrez search (see also Chapter 15 and Chapter 16).
Segmented Nucleotide Sets
A segmented nucleotide entry is a set of non-contiguous sequences that has a defined order and orientation. For example, a genomic DNA segmented set could include encoding exons along with fragments of their flanking introns. An example of an mRNA segmented pair of records would be the 5′ and 3′ ends of an mRNA where the middle region has not been sequenced. To import nucleotides in a segmented set, each individual sequence must be in FASTA format with an appropriate definition line, and all sequences should be in the same file.
High-Throughput Genomic Sequences
Genome Sequencing Centers use automated sequencing machines to rapidly produce large quantities of “unfinished” DNA sequence, called high-throughput genomic sequence (HTGS). These sequences are not usually annotated with any features, such as coding regions, at all, and in the initial phases are not of high (“finished”) quality.
The sequencing machines produce intensity traces for the four fluorescent dyes that correspond to the four bases adenine, cytosine, guanine, and thymine. Software such as PHRED and PHRAP convert these raw traces into the sequence letters A, C, G, or T. PHRED is a base-calling program that “reads” the sequences of the DNA fragments and produces a quality score. With multiple overlapping reads to work on, PHRAP assembles the DNA fragments using the quality scores of PHRED, itself producing a quality score for each base. The resulting file, which PHRAP outputs in “.ace” format, consists of the sequence itself plus the associated quality scores. Sequin can use these files as input and assemble valid GenBank records from them. Further information on using Sequin to prepare a HTGS record can be found here.
Feature Tables
Some Genome Centers now analyze their sequences and record the base positions of a number of sequence features such as the gene, mRNA, or coding regions. Sequin can capture this information and include it in a GenBank submission as long as it is formatted correctly in a feature table. Sequin can read a simple, five-column, tab-delimited file in which the first and second columns are the start and stop locations of the feature, respectively, the third column is the type of feature (the feature key—gene, mRNA, CDS, etc.), the fourth column is the qualifier name (e.g., “product”), and the fifth the qualifier value (e.g., the name of the protein or gene). The features for an entire bacterial genome can be read in seconds using this format. A sample features table is shown below.
>Feature sde3g
240 4048 gene
gene SDE3
240 1361 mRNA
1450 1614
1730 3184
3275 4048
product RNA helicase SDE3
579 1361 CDS
1450 1614
1730 3184
3275 3880
product RNA helicase SDE3
Alignments
Population, phylogenetic, and mutation studies all involve the alignment of a number of sequences with each other so that regions of sequence similarity are emphasized. Sometimes it is necessary to introduce gaps into the sequences to give the best alignment. Sequin reads several output formats from sequence and phylogenetic analysis programs, including PHYLIP, NEXUS, PAUP, or FASTA+GAP.
The submitted sequence alignment represents the relationship between sequences. This inferred relationship allows Sequin to propagate features annotated on one sequence to the equivalent positions on the remaining sequences in the alignment. Feature propagation is one of the many editing functions possible in Sequin. Using this tool significantly reduces the time required to annotate an alignment submission.
Automated Submission
tbl2asn is a program that automates the submission of sequence records to GenBank. It uses many of the same functions as Sequin but is driven entirely by data files, and records need no additional manual editing before submission. Entire genomes, consisting of many chromosomes with feature annotation, can be processed in seconds using this method.
Packaging the Submissions
Sequences given to Sequin in the input data formats described in this chapter are retained within Sequin memory, allowing them to be manipulated in real time. For example, for submission to GenBank, the sequence is transformed from the Sequin internal structure to Abstract Syntax Notation 1 (ASN.1), the data description language in which GenBank records are stored. This is the format transmitted over the Internet when submitting to GenBank. Sequin can also output information in other formats, such as GenBank flatfile or XML, for saving to a local file.
Most sequence submissions are packaged into a BioseqSet, which contains one or more sequences (Bioseqs), along with supporting information that has been included by the submitter, such as source organism, type of molecule, sequence length, and so on (Figure 1). There are different classes of BioseqSets; thus, a simple single nucleotide submission is called a nuc-prot set (a BioseqSet of class nuc-prot) containing the nucleotide and protein Bioseqs. Similarly, population, phylogenetic, and mutation sequences, along with alignments, are packaged into BioseqSets of classes pop-set, phy-set, and mut-set, respectively. The alignment information is extracted into a Seq-align, which is packaged as annotation (Seq-annot) associated with the BioseqSet. In the case of PHRAP quality scores, these are converted into a Seq-graph, which, similar to alignment information, is packaged in a Seq-annot; however, in this case it is associated with the nucleotide sequence and not the higher-level BioseqSet. The Seq-graph of PHRAP scores can be displayed in Sequin's Graphical view.
Features are usually packaged on the sequence indicated by their location. For example, the gene feature is packaged on the nucleotide Bioseq, and a protein feature is packaged on the protein Bioseq. Proteins are real sequences, and features such as mature peptides are annotated on the proteins in protein coordinates (although they can be mapped to nucleotide coordinates for display in a GenBank flatfile). A CDS (coding region) feature location points to the nucleotide, but the feature product points to the protein. For historical reasons, the CDS is usually packaged on the nuc-prot set instead of on the nucleotide sequence.
Viewing and Editing the Sequences
After the record has been constructed, the features can be viewed in a variety of display formats (Table 1). These include the traditional GenBank or GenPept flatfiles, a graphical overview, the feature spans displayed over the actual sequence letters, and ASN.1. These formats are generated by components of the NCBI Toolkit.
Table 1
The display formats available in Sequin.
The different format generators all work independently from one another. When Sequin starts up, it registers a set of function procedures used to generate each display format. While issuing Sequin commands during manipulation of the sequence, appropriate messages (for example, “generate the view from the internal sequence record”, “highlight this feature”, “export the view to the clipboard”, etc.) are sent to the viewer by calling one of these procedures. Separate lists of registered formats are maintained for nucleotide, protein, and genome record types.
Just as the different format generators do not need to know about each other, Sequin's viewer windows do not need to know about other Sequin viewer or editor windows that are active at the same time. When editing a sequence, the user may have several different views of the same sequence open at the same time (for example, a GenBank flatfile and a graphical view). Clicking on a feature in the graphical view will select the same feature in the GenBank flatfile, and double-clicking on a feature launches the specific editor for that feature. This type of communication between different windows is orchestrated by the NCBI Toolkit's object manager.
The Sequence Editor
The sequence editor is used like a text editor, with new sequence added at the position of the cursor. Furthermore, the sequence editor automatically adjusts the biological feature intervals as editing proceeds. For example, if 60 bases are pasted or typed onto the 5′ end of a sequence record, the sequence editor will shift all the features by 60 bases. This means that interval correction does not need to be done by hand. Prior to Sequin, it was usually easier to resubmit from scratch than to edit all of the feature intervals manually.
The Feature Editors
Feature editor windows have a common structure, organized by tabs (Figures 2–4). The first tab is for elements specific to the given feature (Figure 2). For example, in a coding region editor, the first tab has controls for entering the coding region and reading frame. The second tab is for elements common to all sequence features (Figure 3). These include an exception flag, which allows explanations to be given for unusual events (e.g., RNA editing or non-consensus splice sites) and a comment (a free-text statement shown as “/note” in the flatfile). The last tab is a location spreadsheet allowing multiple feature intervals to be entered (Figure 4). For a coding region, these would reflect the boundaries of the exons used to encode the protein.

Figure 2
The first feature editor tab, Coding Region, allows entry of information specific to the given type of feature.

Figure 3
The Properties tab is for entry of information common to all types of features.
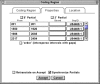
Figure 4
The Location tab has a spreadsheet for entry of feature locations.
Computational Functions of Sequin
Sequin combines several NCBI Toolkit functions to perform many useful computations on the data in Sequin's memory.
Automatic Annotation of Coding Regions
When a nucleotide is submitted to GenBank using Sequin, it is essential to give the name of the source organism. The submitter is also strongly encouraged to supply the translated protein sequence(s) for the nucleotide.
Supplying the organism name allows Sequin to automatically find and use the correct genetic code for translating the nucleotide sequence to protein for the most frequently sequenced organisms. On the basis of only the genetic code and sequences of the nucleotide and protein products, Sequin will then calculate the location of the protein-coding region(s) on the nucleotide sequence. This is an extremely powerful function of Sequin. The ability to do this automatically, instead of by hand, has made sequence submission much faster and less error-prone.
Sequin uses a reverse translating alignment algorithm, called Suggest Intervals, to locate the protein-coding region(s) on the nucleotide sequence. The algorithm builds a table of the positions of all possible stretches of three amino acids in the protein. It then translates the nucleotide in all six reading frames and searches for a match to one of these triplets. When it finds one, it attempts to extend the match on each side of the initial hit. If the extension hits a mismatch or an intron, it stops. Given these candidate regions of matching, Sequin then tries to find the best set of other identical regions that will generate a complete protein. While doing this, the algorithm takes splice sites into account when deciding where to start an intron in eukaryotic sequences and fuses regions split by a single amino acid mismatch.
Updating Sequence Records
The ability to propagate features through an alignment and the way the sequence editor can adjust feature positions as the sequence is edited are combined in Sequin to provide a simple and automatic method for updating an existing sequence.
The Update Sequence function allows overlapping sequence or a replacement sequence to be included in an existing sequence record. Sequin makes an alignment (using the BLAST functions in the NCBI Toolkit), merges the sequence if necessary, and propagates features onto the new sequence in the new positions. This effectively replaces the old sequence and features. The Update Sequence function is based on the NCBI Toolkit's alignment indexing, which allows Sequin to produce several displays that help the user to confirm that the correct sequence is in fact being processed (Figure 5).

The Validator
The final version of the sequence, complete with all the annotated features, can be checked using the validator. This function checks for consistency and for the presence of required information for submission to GenBank. The validator searches for missing organism information, incorrect coding-region lengths (compared to the submitted protein sequence), internal stop codons in coding regions, mismatched amino acids, and non-consensus splice sites. Double-clicking on an item in the error report launches an editor on the “offending” feature. The NCBI Toolkit has a program (testval), which is a stand-alone version of the validator.
The validator also checks for inconsistency between nucleotide and protein sequences, especially in coding regions, the protein product, and the protein feature on the product. For example, if the coding region is marked as incomplete at the 5′ end, the protein product and protein feature should be marked as incomplete at the amino end. (Unless told otherwise, the CDS editor will automatically synchronize these anomalies, facilitating the correction of this kind of inconsistency.)
Additional checks include ensuring that all features are annotated within the range of the sequence, all feature location intervals are noted on the same DNA strand, tRNA codons conform to the given genetic code, and that there are no duplicate features or different genes with the same names. The validator even checks that the sequence letters are valid for the indicated alphabet (e.g., the letter "E" may appear in proteins but not in nucleotides).
In cases where an exception has been flagged in a feature editor, specific validator tests can be disabled. For example, if the reason given for an exception is “RNA editing”, this turns off CDS translation checking in the validator. Likewise, “ribosomal slippage” disables exon splice checking, and “trans splicing” suppresses the error message that usually appears when feature intervals are indicated on different DNA strands.
Automatic Definition Line Generator
NCBI has a preferred format for the definition line in the GenBank format. It starts with the organism name, then the names of the protein products of coding regions (with the gene name in parentheses), with “complete” or “partial” at the end:
DEFINITION Human T-cell lymphotropic virus type 1 isolate ES-TMD envelope
glycoprotein (env) gene, partial cds.
There is also a standard style for explaining alternative splice products. Sequin's Automatic Definition Line Generator collects CDS, RNA, and exon features in the order that they appear on the nucleotide sequence, finds the relevant genes (usually by location overlap), and prepares a definition line that conforms to GenBank policy.
Recalculation of Multiple CDS Features
In spite of all the safeguards built into Sequin, a submitter sometimes uses an incorrect genetic code for an organism. This means that the protein products of CDS translations may be incorrect. Sequin can retranslate all CDS features with a single command. Even so, if the sequence being edited is large, for example, a whole chromosome, this can be a time-consuming operation. To speed it up, the NCBI Toolkit uses a finite state machine (an efficient pattern search algorithm) for rapid translation. The machine is primed with a given genetic code, and then nucleotide sequence letters are fed into the algorithm one at a time, in the order they appear in the sequence. This allows all six frames (three frames on each strand) to be translated in the least possible time. The Open Reading Frame search in the NCBI Toolkit's tbl2asn program also uses this function.
Advanced Topics
The Special Menu
The Special Menu of Sequin encompasses a powerful set of tools that are available to GenBank and Reference Sequence indexers only. The Special Menu is not available to the public in the standard release because without a thorough understanding of the NCBI Data Model, use of the functions can cause irreparable damage to a record. It allows indexers to globally edit features, qualifiers, or descriptors in all sequences in a record, so that the same correction does not have to be made at each occurrence of the error. For example, all CDS features with internal stop codons can be converted to pseudogenes. Another common error made by submitters is to enter a repeat unit (a rpt_unit; e.g., ATTGG) in the repeat-type field (a rpt_type; e.g., tandem repeat). The Special Menu allows indexing staff to convert rpt_type to rpt_unit throughout the record.
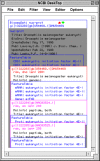
NCBI Desktop Window
Although Sequin has editors for changing specific fields and Special Menu functions for doing bulk changes on several features, it is not possible to anticipate all of the manipulations NCBI indexers might need to do to clean up a problem record. The NCBI Desktop window shows the internal structure of a record (Figure 1), i.e., how Bioseqs are packaged within Bioseq-sets, and where features, alignments, graphs, and descriptors are packaged on the sequences or sets. Objects (sets, sequences, features, etc.) can be dragged out of the record or moved to a different place in the record. Such manipulations could break the validity of the sequence record; therefore, great care must be taken when using it.
For technically adept Sequin users, the Desktop is where additional analysis functions can be added to Sequin without building a complicated user interface. With a feature or sequence selected, items in the Filter menu perform specific analyses on the selected objects. The standard filters include reverse, complement, and reverse complement of a sequence, and reverse complement of a sequence and all of its features. These are needed to repair the occasional record that came in on the wrong strand or in 3′→5′ direction. Adding new filter functions requires adding code to one of the Sequin source code files (one is provided with no other code in it for this purpose) and recompiling the program.
Network Analysis Functions
The functions of Sequin can be expanded by the addition of a configuration file that specifies the URLs for other programs (CGI scripts) available from the Internet. For example, tRNAscan-SE, a program by Sean Eddy and colleagues at Washington University (St. Louis, MO), can be used on sequences in Sequin in this way.
At a minimum, the CGI should be able to read FASTA format and should return either sequence data or the five-column feature table (discussed above) as its result. The external programs that Sequin knows about appear in the Analysis menu. When one of these analyses is triggered, Sequin sends a message to the URL and checks for the result with a timer so that the user can continue to work while the (Web) server is processing the request. The code for the CGI that chaperones data from Sequin to tRNAscan-SE and converts the tRNAscan results to a five-column feature table is in the demo directory of the public NCBI Toolkit.
Network Fetch Functions
As sequencing of complete bacterial genomes and eukaryotic chromosomes becomes more commonplace, the demand to break up long sequences into more manageable bites has increased (although Sequin is perfectly capable of editing these large records). Some genomes at NCBI are thus represented as segmented (or delta) Bioseqs, which are composed of pointers to other raw sequences in GenBank. Obtaining the entire sequence and all of the features requires fetching the individual components from a network service.
The object manager allows Sequin to know about different fetch functions that can be used. When a sequence is needed, these functions will be called until one of them satisfies the request. For example, the lsqfetch configuration file can be edited to point to a directory containing sequence files on a user's disk. The SeqFetch function calls a network service at NCBI to obtain sequences and to look up Accession numbers given gi numbers or gi numbers given Accession numbers.
When used internally by NCBI indexers, Sequin can also fetch records from the DirSub and TMSmart databases. To ensure the confidentiality of pre-released records, this access requires the indexer to have a database password and to be working from a computer within NCBI. For additional protection, the paths to the database scripts are stored in a configuration file and are not encoded in the public Sequin source code.
Conclusion
Sequin has an important dual role as a primary submission tool and as a full-featured sequence record editor. It is designed to be modular on several levels, which simplifies the design and implementation of its components. Sequin sits at the top of the NCBI software Toolkit, relying on many of the underlying components, and thus acts as a quality assurance that these functions are working properly.
- Sequin: A Sequence Submission and Editing Tool - The NCBI HandbookSequin: A Sequence Submission and Editing Tool - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...