

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997.

Retroviral Replication Errors and Genetic Diversity
Retroviral populations evolve rapidly to fit the changing requirements of the environments in which they replicate (Katz and Skalka 1990; Kohlstaedt et al. 1992; Coffin 1993, 1995; Jacobo-Molina et al. 1993; Wain-Hobson 1993). They can expand or alter their tropism, evade the defenses of their hosts, or adapt to survive drugs designed to block their replication. Although errors in replication provide the basis for diversity, in the case of viruses like HIV-1, the diversity seen in patients is the result of large numbers of virions present in the patient and the high rate of replication (Coffin 1995). About one third of the integrants generated by infection by wild-type MLV display readily detectable defects (Shields et al. 1978). Errors can be introduced into the genome at any of a number of stages in the viral replication cycle, and it is not presently clear which stages are the most significant. First, retroviral genomic RNA is a transcript of host RNA polymerase II, which does not have a proofreading function; thus, transcription of viral RNA may have a role in generating diversity. Second, certain types of genetic alterations—insertions, deletions, and apparent template switches—are unlikely to result from errors made by RNA polymerase, and these alterations suggest that RT has a significant, although imperfectly defined, role in generating retroviral diversity. Third, host DNA repair enzymes may be involved in filling gaps or completing plus-strand synthesis, and some of these systems are error-prone as well. Ultimately, however, it is environmental selective pressure that determines the genetic composition of retroviral populations (Ji and Loeb 1994; Bonhoeffer et al. 1995; Coffin 1995).
Populations of HIV-1 appear to be more genetically polymorphic than human T-cell leukemia viruses (HTLVs), and it has been suggested that this results from unusually high error rates during HIV replication (Myers and Pavlikis 1992; Wain-Hobson 1993). However, it now seems likely that the rate at which variants, including drug-resistant variants, arise in HIV-1 is no greater than in other retroviruses but that the high rate of HIV replication provides an increased opportunity for the generation of variants (Coffin 1995). Studies of viral population dynamics in HIV-infected individuals suggest that the rates of viral replication during periods of clinical latency are much higher than what was previously believed (Coffin 1995; Ho et al. 1995; Wei et al. 1995; see also Chapter 11. In particular, it appears that the course of HIV disease involves a dynamic cycle of continuous rounds of new infection, viral replication, and infected cell turnover. This suggests that the end of clinical latency may result from an accumulation of damage to the immune system and offers the renewed hope that agents which reduce this damage by decreasing viral load may increase the duration of latency sufficiently that HIV infection becomes a manageable condition rather than an inevitable prelude to AIDS (Coffin 1995; see also Chapters 11 and 12).
RT Fidelity In Vitro
All RTs show poor fidelity when compared with host DNA polymerases in in vitro systems (Battula and Loeb 1974; Preston et al. 1988; Roberts et al. 1988; Takeuchi et al. 1988; Weber and Grosse 1989; Boyer et al. 1992a; Kati et al. 1992; Bebenek et al. 1993; Patel and Preston 1994; for reviews, see Bebenek and Kunkel 1993; Williams and Loeb 1992). The lack of a proofreading function (Battula and Loeb 1976) probably explains much of this high error rate, although the enzyme's ability to extend mismatched primer termini may contribute to a tendency to incorporate “wrong” nucleotides (Perrino et al. 1989; Bakhanashvili and Hizi 1992; Pulsinelli and Temin 1994). As discussed below, if the tendency of RT to switch templates is paired with its ability to extend mismatches, genetic recombination may be a significant source of replication errors.
The error rates of various RTs have been estimated in many different types of assays. Some of these assays involve measuring misincorporation of a nontemplated triphosphate on a homopolymeric template (Loeb and Kunkel 1982; Takeuchi et al. 1988). Others involve measuring mutation rates at particular sites in a heteropolymeric template; one such assay involves an RT-mediated reversion at an amber codon (Weymouth and Loeb 1978). A larger net can be cast by measuring the rate of forward mutation in a functional gene (Kunkel 1985). In all such assays, the error rate can be significantly affected by altering the parameters of the reaction such as the relative and absolute concentrations of the triphosphates, the monovalent and divalent salt concentrations, the mutation being introduced, and the enzyme being examined (Bebenek et al. 1989, 1993; Roberts et al. 1989; Ricchetti and Buc 1990; for review, see Bebenek and Kunkel 1993). The error rate is quite dependent on sequence context, and “hot spots” where errors occur frequently may differ according to the particular RT used in the assay (Ricchetti and Buc 1990; Bebenek et al. 1993). In addition, error rates may be different on RNA and DNA templates (Boyer et al. 1992a; Yu and Goodman 1992). As a consequence, it is not possible to define the error rate as a single number. These studies, taken together, do suggest that the overall error rate is high and that the misincorporation rate of most RTs in vitro, under physiological conditions, is on the order of 10–4 errors per base incorporated. This rate would translate into an error rate in viruses of about one per genome in a reverse transcription cycle, even without considering errors made by host RNA polymerase. Thus, although factors in cells may alter rates and some studies suggest that somewhat lower error rates occur in vivo (discussed below), the in vitro data lead to the generally accepted impression that the average retroviral DNA genome differs from its parent by at least one mutation and provide evidence to justify the argument that retroviruses consist of “quasi-species” or “swarms” (Wain-Hobson 1993).
Replication Errors In Vivo
When the rates at which mutations accumulate in retroviral and host genomes are compared, the viral genomes are found to evolve at rates perhaps a million-fold higher than the genomes of their hosts (Gojobori and Yokoyama 1985). Several groups have attempted to determine the actual rate at which mutations accumulate during retroviral replication by limiting (or defining) the number of infectious cycles and quantifying the appearance of genetic changes. Experiments have been carried out with both replication-competent viruses and replication-defective viral vectors. Both approaches have inherent limitations. Nonlethal mutations are favored in the first approach and phenotypically detectable mutations (i.e., changes in reporter genes) in the latter. Experiments that involve a single round of infection usually involve a defective retroviral vector and a complementary helper cell that produces, in trans, essential viral proteins deleted from the vector. Viral vectors of this type are described in Chapter 9.
Depending on whether the expression of a suppressed gene or the reversion of an amber codon was monitored, the mutation rates in a single cycle of SNV replication have been estimated at 5 × 10–3 per base pair and 2 × 10–5 per base pair, respectively (Dougherty and Temin 1986, 1988). In assays where the point mutation rate was examined by comparing the RNase T1 oligonucleotide fingerprinting patterns of the RNAs of the progeny of an infectious MLV after a single round of replication, the substitution rate was 2 × 10–5 per base pair (Monk et al. 1992). A screen of several discrete regions of the RSV genome for virus that had un- dergone a single cycle of replication yielded an apparent substitution rate of 1.4 × 10–4 per base pair (Leider et al. 1988). In all of these studies, it is unclear whether the measured error rates, which are significantly higher than those of the host DNA replication machinery, are due to errors by host RNA polymerase or RT. A comparison of the accumulation of mutations by two different viruses copying the same sequence during a single replication cycle indicated that SNV replication was somewhat less error prone than that of bovine leukemia virus (Mansky and Temin 1994). The concordance between error rates measured for purified RT in vitro and for retroviral replication in vivo varies from “very good” to “significantly more errors in vitro,” depending on which studies are compared. Although the differences in experimental approach or error detection may account for much of the discrepancy, it is entirely possible that RT fidelity may be enhanced in vivo, possibly by viral proteins or host factors that enhance fidelity and processivity.
There is limited evidence both for and against the possibility that features of the template that impede RT in vitro may also cause replication errors in vivo (Alford and Belmont 1990; Pathak and Temin 1992; Varela-Echavarria et al. 1992; Jones et al. 1994). The tendency of RT to dissociate from its template may affect both the rate and position of the RT-mediated recombination that takes place via template switching between two copackaged genomic RNAs. In vitro, forced template switching can occur at very high rates (Luo and Taylor 1990; Buiser et al. 1991; Garces and Wittek 1991; DeStefano et al. 1992b, 1994a; Peliska and Benkovic 1992).
An interesting phenomenon noted in the analysis of mutants arising during retroviral replication is the formation of blocks of mutations that are clustered in small portions of the genome. Retroviral “hypermutations” have been observed in the genomes of SNV, HIV-1, and caprine arthritis encephalitis virus (CAEV), consisting of clustered G to A, sometimes up to 30% of all Gs in a localized region (Pathak and Temin 1990a; Vartanian et al. 1991; Borman et al. 1995; Wain-Hobson et al. 1995). These substitutions most commonly arise in the context of G-A dinucleotides, which has led to the proposal that they arise via dislocation mutagenesis (slippage of the primer strand during DNA synthesis) or at the ends of runs of Gs (Vartanian et al. 1991; Fitzgibbon et al. 1993). It has also been suggested that these hypermutations may arise from incorporation by a variant RT prone to high error rates or from incorporation by RT under periods of stress—such as low triphosphate levels—when error rates might be very high (Pathak and Temin 1990a; Vartanian et al. 1991, 1994; Varela-Echavarria et al. 1992). It is possible that these clustered errors might arise by other mechanisms, for example, by host error-prone repair machinery acting on short patches of duplex DNA. A to G hypermutations were observed in an inverted repeat region of an avian retroviral recombinant. Such mutations may have resulted from the action of host double-stranded RNA adenosine deaminase, an enzyme that catalyzes A to I to G changes and has been implicated in A to G hypermutations in other RNA viruses (Cattaneo 1994; Hajjar and Linial 1995).
Genetic Recombination
Mechanisms of Recombination
Since retroviruses ordinarily copackage two identical, or nearly identical, RNAs, the genetic consequences of using only one versus portions of both of the RNAs to template DNA synthesis are usually the same. However, under certain conditions, two genetically distinct RNAs can be copackaged. Portions of each template can be used to generate a single DNA, producing a recombinant. Although recombination could, in theory, occur between two viral RNAs introduced into a single cell by different virions, this does not appear to occur at a measurable frequency. Recombination does occur at high frequency in virions produced by a single cell that has been coinfected by two different viruses (Wyke et al. 1975; Clavel et al. 1989), demonstrating that recombination requires copackaging of two viral genomes into the same particle (Hu and Temin 1990a,b; Stuhlmann and Berg 1992). During reverse transcription in such heterozygous virion particles, product DNAs can be generated that contain portions of each of the two genomic sequences in one molecule. It has been calculated that during a single cycle of reverse transcription, homologous recombination between two genetic markers 1 kb apart occurs in about 4% of the DNAs produced in heterozygous virions (Hu and Temin 1990a). The rate of nonhomologous recombination is significantly lower—perhaps only 1/100 to 1/1000 as frequent—and also depends on the extent of copackaging of heterologous RNA (Zhang and Temin 1993b) (see discussion of transduction below).
Two models have been proposed to explain genetic recombination during reverse transcription. In the copy choice model, genetic recombination occurs during the first, or minus-strand, DNA synthesis; in the strand displacement-assimilation model, recombination occurs during plus-strand DNA synthesis. These two models are not mutually exclusive, and there is experimental support for both. However, it is likely that most retroviral recombination occurs during minus-strand synthesis.
In the copy choice model, recombination occurs when the growing DNA chain switches from one RNA template to another during minus-strand DNA synthesis. The original “forced copy choice” version of this model proposes that recombinogenic template switching occurs when a break in template RNA forces RT to seek a homologous template region on a copackaged RNA (Coffin 1979). A prediction of this model is that conditions which increase RNA damage should stimulate recombination, but attempts to demonstrate this with γ-irradiation were ambiguous (Hu and Temin 1992). More recently, the copy choice model has been broadened to include template switching from regions of RNA that are unbroken (Xu and Boeke 1987). Evidence for template switching from internal regions of RNA templates has come from both in vitro reactions and viral genetic studies (DeStefano et al. 1994a). Some regions, including remnants of polylinkers whose secondary structures may induce pausing by RT, are recombinational hot spots in viral vectors (Pathak and Temin 1990b; Jones et al. 1994).
The strand-displacement assimilation model was originally proposed on the basis of electron microscopic observations of H-branched structures that may have been recombination intermediates (Junghans et al. 1982b). As noted above, plus-strand synthesis is discontinuous and initiated at multiple points in at least some retroviruses (Boone and Skalka 1981b; Kung et al. 1981; Hsu and Taylor 1982; Miller et al. 1995). According to this model, plus-strand DNA synthesis can continue far enough that the 5′ends of the nascent plus strands are displaced by 3′ends of the adjacent plus strands. The displaced portions would then be free to base pair with the other minus-strand DNA in the virion. Genetic recombination would result if the displaced plus-strand fragment was incorporated into the viral genome. This mechanism is supported by observations that in reactions in vitro, RT can mediate DNA unwinding or strand displacement (Collett et al. 1978; Whiting and Champoux 1994); recent results suggest that concomitant DNA synthesis may be required for strand displacement (Whiting and Champoux 1994). Note that for viruses which synthesize plus strands discontinuously, two completed minus strands are not necessary for strand-displacement assimilation. However, whereas recombination during minus-strand synthesis requires only that two genetically distinct RNAs be copackaged, recombination during plus-strand synthesis requires that two minus-strand DNA copies of the region where recombination will occur must be present.
Is Retroviral Recombination Mutagenic?
The first strand transfer reaction and retroviral recombination—especially forced copy choice recombination—are mechanistically related. Various researchers have suggested that strand transfer is an error-prone process (Peliska and Benkovic 1992, 1994; Darlix et al. 1993; Patel and Preston 1994) or that it is an important mechanism in the generation of retroviral genetic diversity (Temin 1993). It has been suggested that the ability of RT to perform strand transfers, which is a requirement for viral replication, predisposes the enzyme to generate errors (Coffin 1979; Temin 1993). In reconstituted reactions in vitro, RT can incorporate additional nontemplated nucleotides when it reaches the end of a template (Peliska and Benkovic 1992; Patel and Preston 1994). RTs are known to extend mismatches more readily than other DNA polymerases (Perrino et al. 1989; Bakhanashvili and Hizi 1992; Yu and Goodman 1992). It has been suggested that forced copy-choice-type recombination might be mutagenic if RT adds a nontemplated base when stalled at a broken template end and then extends the DNA from this incorrect nucleotide following transfer to the second template (Peliska and Benkovic 1992; Patel and Preston 1994). An in vitro system designed to test the fidelity of forced copy choice recombination showed no evidence of frameshift mutations, but there was misincorporation, presumably from nontemplated base additions, in 5–20% of the strand transfer products (Peliska and Benkovic 1994). A similar rate of errors was observed during first strand transfer by MLV during intracellular reverse transcription (Kulpa et al. 1997). On the basis of the frequency of blunt-end addition in vitro (30–50%) and the rate of retroviral recombination, it has been suggested that in a single cycle of replication, 5–10% of all viruses could carry mutations generated by this mechanism (Patel and Preston 1994).
DNA sequences at recombination junctions generated during retroviral replication appear to rule out the possibility that recombination generates mutations at this high rate. No mutations were found in any of 30 recombinant MLV-based vectors in the region where recombination had occurred (Zhang and Temin 1994). This observation does not exclude the possibility that recombination may be mutagenic under certain circumstances; for example, there may be sequence-specific effects on the rate of mutation, and other sorts of mutations, for example, short insertions, could arise through template switching (Pathak and Temin 1990b). Moreover, whether or not recombination is mutagenic may depend on whether recombination occurs at the end of a broken template or from an internal template position. RT clearly can extend mismatched primer termini, even in vivo (Pulsinelli and Temin 1994; Das and Berkhout 1995). In experiments where the downstream edge of the PBS was mutated, no decrease in viral titer was observed, and RT could efficiently perform the second strand transfer and extend even a three-base mismatch (Pulsinelli and Temin 1994). This observation suggests that the presence of a mismatch would not prevent recombination from occurring and that recombination would be mutagenic if it were common for nontemplated bases to be added when DNA synthesis reached the end of a template. However, a second study observed that whereas mismatches that arose during strand transfer could be extended, even a single template-primer mismatch decreased viral titer (Kulpa et al. 1997). The fact that recombination does not generally appear to be mutagenic in vivo suggests that terminal nucleotide addition occurs less frequently in vivo than in vitro (perhaps because template switching is more rapid than nontemplated base addition) and/or that recombination occurs more frequently from the internal regions of templates than at broken template ends. It may also be that when mismatches arise, strand transfer may fail and DNA synthesis then be aborted, thus leading to the observed underrepresentation of error-prone transfer products among complexed DNAs.
Transduction of Cellular Genes: Oncogene Capture
The genomes of acute transforming retroviral genomes contain cellular proto-oncogenes embedded within flanking retroviral sequences. The structures of acute transforming viruses vary widely, but there are several invariant features. Segments required in cis for viral replication are always retained: the LTRs, the PBS, and the PPT are present because they are required for reverse transcription of the genome. The RNA packaging signals are retained to permit the encapsulation of the RNA into virions. Portions of the gag, pol, and env genes may or may not be present. These genes are often deleted and replaced with sequences derived from the host genome; as a rule, the inserted sequences consist of spliced, exonic sequences rather than genomic sequences containing introns, and the inserts generally lie in the same transcriptional orientation as the viral genome. These requirements are similar to what is required in a defective retroviral vector (for a more detailed description of these features, see Chapter 9. The structures of the genomes of acute transforming retroviruses suggest that a series of rare events, probably including aberrant reverse transcription, are involved in the generation of oncogene-containing retroviruses (Goldfarb and Weinberg 1981a,b; Swanstrom et al. 1983). One model for retroviral transduction of cellular genes (currently the most popular one) is outlined here and illustrated in Figure 9. This model is undoubtedly simplistic, since the structures of acute transforming viruses vary and some are more complex than this model predicts. However, the structures of some of the extant transforming viral genomes provide strong support for the model. The following are the steps in transduction:
- 1.
A retrovirus integrates upstream of the gene to be transduced.
- 2.
Readthrough transcription from the provirus generates a large RNA containing downstream cellular sequences. Alternatively, deletion in the DNA could directly join viral and cellular sequences, and transcription would produce the chimeric viral/cellular RNA.
- 3.
The chimeric RNA is packaged into virions as a heterodimer along with RNA from a wild-type helper. In some cases, RNA splicing contributes to the generation of a chimeric RNA that can be readily packaged.
- 4.
Nonhomologous template switches between copackaged helper RNA and the chimeric RNA during reverse transcription introduce the requisite viral sequences downstream from the cellular gene. The resulting DNA genome can be integrated normally and serves to generate viral RNAs that can be replicated efficiently, producing a high-titer acutely transforming virus.
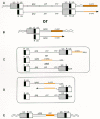
This and related models postulate that RNA intermediates and reverse transcription are crucial for the transduction of cellular genes by retroviruses. Perhaps the strongest evidence that mRNAs are directly involved in transduction is the fact that poly(A) stretches are present at the host–3′viral junctions in viruses carrying portions of the erb-B and c-fps genes (Huang et al. 1986; Raines et al. 1988). Since these poly(A) runs are not present in the viral genome or in host DNA but only in host-derived mRNAs, their presence strongly implicates such mRNAs as donors to the genomes of these viruses. The simplest mechanism accounting for their presence is an RT-mediated transfer of DNA synthesis from viral 3′sequences to the poly(A) sequences of a packaged chimeric RNA. Junctions between a captured oncogene and the 3′sequence sometimes occur in short segments where there is fortuitous homology, but regions of homology are not required (Stavnezer et al. 1989; Zhang and Temin 1993a,b).
Variations of this model differ in how the 5′virus oncogene junction is established. It is possible that a deletion in chromosomal DNA joins the 5′portion of an integrated provirus to the gene. However, such deletions have not been directly observed and this mecha- nism is likely to be exceedingly rare. A chimeric RNA can be produced from an intact viral genome integrated upstream of an oncogene if the host RNA polymerase reads through the LTR. Such readthrough events apparently occur relatively frequently; perhaps 15% of viral transcripts fail to terminate in the LTR and contain downstream host sequences. Such RNAs can be packaged efficiently and reverse-transcribed (Herman and Coffin 1986; Swain and Coffin 1989, 1992).
There is direct support for a readthrough RNA model in the generation, in ALV-infected chickens, of acutely transforming retroviruses that carry a portion of the EGF receptor (erb-B) gene. As predicted in the model, cellular transformation is correlated with the integration of an intact ALV provirus upstream of the Erb-B sequences that will be captured. In the case of Erb-B, which is a receptor tyrosine kinase, the provirus is inserted into an intron upstream of the kinase domain. Readthrough transcription generates a chimeric viral/cellular RNA. A cryptic splice donor in the env gene is joined to a splice acceptor site in the erb-B gene, generating a message that codes for an in-frame gene fusion product (Nilsen et al. 1985; Raines et al. 1988). Transformed cells can be isolated from infected birds that produce this spliced RNA but do not yet produce an infectious, acutely transforming, virus. Not surprisingly, these cells do eventually give rise to acutely transforming virus, and the viruses that are isolated appear to have incorporated 3′viral sequences, presumably via reverse transcription. Although these data do not rule out the possibility that retroviruses use other mechanisms (e.g., DNA-mediated recombination) to capture cellular oncogenes, they do provide evidence that an RNA readthrough mechanism is actually used in the capture of oncogenes in vivo.
The capture of an oncogene by a retrovirus is a rare event in an infected animal; it has not been possible to develop tissue culture systems in which such events can be demonstrated. It is possible, however, to prepare model systems that employ retroviral vectors that carry selectable markers to study events similar to the events thought to be involved in oncogene capture. The initial experiments were done with fusions of the 5′viral sequences and a selectable marker. These constructions were intended to mimic the kinds of products that would be produced by a chromosomal deletion. It is possible to select for the reconstruction of the 3′viral sequences by recombination with helper virus genomes in such experiments (Goldfarb and Weinberg 1981a,b; Goff et al. 1982; Stuhlmann et al. 1990). In these studies, each transduction event could be shown to arise by an independent recombination between the chimeric RNA and the helper. The chimeric 5′viral/ selectable marker gene can capture 3′viral sequences in a single round of retroviral replication at 0.1–1% the rate of homologous retroviral replication (Zhang and Temin 1993b). Some of the progeny virus contained deletions and insertions, suggesting that numerous template switches may have occurred during reverse transcription. The formation of some experimental transductants appears to have involved aberrant reverse transcription during both plus-strand and minus-strand synthesis (Zhang and Temin 1993a).
Transduction has also been studied with intermediates that required the formation of both 5′and 3′junctions for successful transfer. For example, Swain and Coffin (1992) placed a selectable marker downstream from a 2-LTR virus, so that readthrough transcripts would include selectable marker gene sequences. To increase the probability of incorporating the selectable sequences into a single RNA, a transcriptional termination mutation was introduced into the viral polyadenylation sequence in the LTR just upstream of the reporter gene. Cells expressing this viral readthrough RNA were infected with replication-competent virus, and fresh cells were infected with the resulting virions. The structure of the resulting proviruses in this and other studies demonstrated that reverse transcription of readthrough transcripts can recreate the entire transduction process in a single cycle of retroviral replication.
A competing model for oncogene transduction proposes that the whole process takes place at the DNA level (Goodrich and Duesberg 1988, 1990a,b). DNA-mediated transduction might involve a second retrovirus integrating downstream from the gene to be transduced, and subsequent deletions of cellular sequences would link the oncogene to the flanking viral sequences. Such purely DNA-based transduction models have not been commonly proposed, and the experimental support for such ideas is not strong, but the possibility that different acutely transforming retroviral genomes were generated by this or other mechanisms cannot be ruled out.
Clearly, acute transforming retroviruses are not always derived in a single cycle of replication. The point mutations found in some transforming viruses probably arose during subsequent rounds of replication, and the presence of VL30 sequences flanking the ras oncogene in three separate acute transforming retroviruses derived in rats suggests that the generation of the recombinant sequences in these viruses was at least a two-step process; however, it is unclear whether incorporation of ras into the retrovirus-like endogenous VL30 sequences preceded recombination between retroviral and VL30 sequences or vice versa (Makris et al. 1993).
All of the above models start with retroviral integration adjacent to the sequences to be transduced as was proposed long ago (Swanstrom et al. 1983). However, it is also conceivable that both the 3′and the 5′viral ends of acute transforming viruses might be acquired by aberrant reverse transcription of normal cellular RNAs that were adventitiously packaged into virions (Stuhlmann et al. 1990; Hajjar and Linial 1993). Even RNAs already present in the target cell might be used as templates (Olsen et al. 1990). An RSV mutant that packages host RNA more efficiently than do wild-type viruses has been described (Linial et al. 1978), and such mutants can mediate transduction of RNAs that contain no viral sequences.
- Special Biological Features of Reverse Transcription - RetrovirusesSpecial Biological Features of Reverse Transcription - Retroviruses
Your browsing activity is empty.
Activity recording is turned off.
See more...