As discussed in Chapter 2, it is the product of the retroviral env gene that allows retroviruses to attach to target cells and penetrate the cell membrane. All env genes occupy a common genomic location downstream from the gag and pol genes, they are expressed via spliced mRNAs, and they have an organization similar to that of precursor proteins, with several common structural features. Nevertheless, they exhibit considerable sequence differences from one another. The mature product, which appears as a knobbed spike on the surface of the virion, comprises the SU and TM proteins, in which the SU domains carry the receptor-binding function. It is the unique ligand-binding domain present within the three-dimensional structure of SU that defines the receptor to which the virus binds and thus the tissue specificity/host range of the virus. In the ASLVs, closely related env sequences recognize distinct receptors; the regions that vary between strains and are implicated in receptor binding are present in a background of relatively constant sequences. The envelope glycoproteins of MLVs resemble those of ASLVs in exhibiting sequence variations that give rise to differences in receptor recognition. In the case of other viruses, such as HIV, considerable variation in SU occurs that can alter both the antigenicity of the protein and the tropism of the virus for specific cell types but, nevertheless, conserves specificity for a single receptor molecule.
General Features of Retroviral Glycoprotein Physical Structure
Multimerization of Env
On the surface of the virion, retroviral glycoproteins are arranged in oligomeric complexes that contain three or four SU-TM heterodimers. This oligomeric structure, which probably provides the mechanical rigidity needed for the complex to assume the knobbed spike structure seen in the electron microscope, forms in the endoplasmic reticulum shortly after translation of the glycosylated Env precursor (Einfeld and Hunter 1988; Chakrabarti et al. 1990; Earl et al. 1991). For the ASLV and MLV Env proteins, the oligomer appears to be a trimer of SU-TM heterodimers (Einfeld and Hunter 1988; Kamps et al. 1991), whereas for the HIV and SIV Env proteins, dimer, trimer, and tetrameric forms have been described (Schawaller et al. 1989; Chakrabarti et al. 1990; Rey et al. 1990; Weiss et al. 1990; Doms et al. 1991). A definition of the molecular form of this oligomer may require three-dimensional structure information.
Although the molecular interactions involved in formation of Env oligomers are poorly understood in the absence of structural detail, it is clear from mutagenic analyses that the domains involved in oligomerization are located within the extracellular domain of the precursor; soluble truncated forms of the ASLV, MLV, and HIV glycoproteins have been shown to form oligomers (Einfeld and Hunter 1988; Tucker et al. 1991; Earl and Moss 1993). Moreover, oligomers of the ASLV and HIV Env proteins appear to form through interactions localized to the TM protein (Einfeld and Hunter 1988; Earl and Moss 1993). In the case of ASLV, the TM protein, expressed independently of SU, forms an oligomer on its own, whereas SU remains monomeric when expressed in the absence of TM (Einfeld and Hunter 1994). Env proteins of HIV-2 and SIV are also oligomeric and share a functionally conserved assembly domain with that of HIV-1 since mixed oligomeric structures can form (Doms et al. 1990).
Oligomerization of Env precursors appears to be important for intracellular transport of the nascent protein, presumably by allowing the sequestration of hydrophobic sequences within the core of the structure. It may also be important for initiating fusion, since both HIV and SIV glycoprotein complexes demonstrate multimeric CD4 binding (Earl et al. 1992), and binding of more than one molecule of CD4/oligomer appears to be necessary for gp120 dissociation and neutralization (Layne et al. 1990; Moore et al. 1991b).
Glycosylation
As with other carbohydrate-modified proteins, retroviral Env proteins are cotranslationally glycosylated by cellular enzymes in the lumen of the rough endoplasmic reticulum that attach mannose-rich oligosaccharides to the asparagine in Asn-X-Ser or Asn-X-Thr motifs (for review, see Hunter and Swanstrom 1990). The number and distribution of N-linked glycosylation sites vary widely between different retroviruses. HIV-1 has as many as 30 of the canonical Asn-X-Ser/Thr oligosaccharide addition sites, with the majority (25) located in gp120. MMTV, on the other hand, has only 4 potential N-linked glycosylation sites, and these are distributed evenly between gp52 and gp37. For the most part, there is little conservation of the positions at which carbohydrate is added among different groups of retroviruses. However, within a particular retroviral group, highly conserved carbohydrate residues can be observed. These may reflect a specific requirement for carbohydrate in the structure/function of the glycoprotein.
The role of carbohydrate side chains in glycoprotein biosynthesis, transport, and stability has been a point of much discussion. Oligosaccharide side chains themselves probably do not act as signals for transport to the cell surface, but they may at least in some cases provide the hydrophilicity required to drive or stabilize correct protein folding, which in turn may be a prerequisite for protein transport and protein function. Synthesis of the HIV-1 glycoprotein in the presence of tunicamycin, an inhibitor of glycosylation, results in a protein that is unable to fold correctly or bind CD4, whereas enzymatic deglycosylation of fully processed secreted gp120/160 causes only minimal changes in receptor binding (Fenouillet et al. 1989; Li et al. 1993). It is likely that carbohydrate, through masking of susceptible residues, enhances stability of the glycoprotein by providing protection from proteolytic enzymes. This masking may also reduce the immunogenicity of the protein, presumably by preventing immunoreactive cells from interacting with polypeptide epitopes (Elder et al. 1986).
Although the bulk of the discussion above has concentrated on N-linked oligosaccharides, additional experiments have indicated that both MLV and HIV Env proteins are also modified by the addition of O-linked sugars (Pinter and Honnen 1988; Hansen et al. 1990; Bernstein et al. 1994). The role and requirement for this posttranslational modification are presently unknown.
Structural Analysis of Env Proteins
Despite efforts to determine a three-dimensional structure for the HIV glycoprotein, this critical information has not been forthcoming, and thus a variety of indirect approaches have been taken to elucidate structural aspects of retroviral Env proteins.
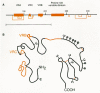
A high degree of variability in the nucleotide sequence of env genes and a preference for changes that alter the amino acid sequence are seen in the primate lentiviruses. Thus, a comparison of HIV-1 isolates led to the identification of five variable regions and four constant regions in gp120, designated V1–V5 and C1–C4 (Fig. 6A) (Starcich et al. 1986; Willey et al. 1986; Modrow et al. 1987), and a similar pattern of variable regions is present in HIV-2 and SIV (Smith et al. 1988). A disulfide map of the cysteines in gp120 has been obtained for a secreted form of the HIV-1 gp120 molecule (Leonard et al. 1990). This map shows that the 18 cysteine residues form nine intrachain disulfide bonds (Fig. 6A). These bonds segregate regions of the protein that have been further delineated by sequence analysis or function. One disulfide loop contains the V1 and V2 regions which are separated from each other by two additional disulfide bonds. The V3 and V4 loops are also delimited by disulfide bonds (Fig. 6B). The sequence immediately downstream from V4, which includes the C4 region thought to be involved in CD4 binding (see below), forms an adjacent loop in which the V4/C4 relationship is superficially similar to that between V1 and V2. Additional disulfide bonds are present in conserved regions amino-terminal to V1/V2 and between V1/V2 and V3 (Fig. 6B). The Env proteins of SIV and HIV-2 isolates have 20 conserved cysteines. The positions of these residues are analogous to those of HIV-1 with the extra pair of cysteines located in the V1/V2 region, suggesting that a similar disulfide network could form (Gregory et al. 1991; Hoxie 1991).

The positions of cysteine residues are generally conserved in the SU proteins of related retroviruses, even though the sequences of the proteins may differ significantly. MLV SU proteins which differ from one another in their receptor specificity have 16 conserved cysteine residues (Fig. 7A). Four additional cysteines are common to ecotropic viruses and occur in an amino-terminal variable region that is involved in receptor binding as discussed below (Ott et al. 1990; Battini et al. 1992; Linder et al. 1994). The disulfide map of MLV gp70 resembles that of gp120 in that the disulfide bonds generally involve cysteines reasonably close to one another in the primary sequence. There are no disulfides linking the amino-terminal and carboxy-terminal regions to one another in either viral SU protein (Figs. 6B and 7B).
Structure-Function of Retroviral Env Proteins
When an enveloped virus encounters a susceptible cell, the earliest steps in the infectious process are designed to bring about fusion of the cellular and viral membranes. This event, which introduces the core of the virus into the cytoplasm of the cell, is mediated by a specific interaction between a glycoprotein on the surface of the virus and a specific receptor (often a glycoprotein) on the surface of the cell. The interaction between the two molecules is followed by a conformational change in the viral glycoprotein, which in turn causes the viral and cellular membranes to be brought sufficiently close together for the membranes to fuse. These processes—receptor binding, structural rearrangement, and membrane fusion—are poorly understood for retroviruses. A primary problem is that a complete detailed three-dimensional structure is lacking for any retroviral glycoprotein.
Much of the current thinking on the mechanisms by which retroviruses enter cells is based on influenza virus. The advantage of the influenza model is that there is considerable structural and functional information. Although it is true that important differences in the details of the process exist, it is also quite likely that there is a considerable similarity in the mechanism(s) that the two viral families use to enter cells. For this reason, we present here an overview of influenza virus entry.
The Influenza Model
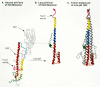
Influenza viruses contain, on their surfaces, a trimeric molecule called hemagglutinin (HA), which is the functional equivalent of the Env glycoprotein of retroviruses. The three-dimensional structure of hemagglutinin is known in considerable detail (see Fig. 8A) (Wilson et al. 1981; Bullough et al. 1994). Like the Env glycoprotein of retroviruses, hemagglutinin is composed of two dissimilar subunits. The larger of these two subunits, HA1, is the functional equivalent of SU, and the smaller, HA2, which is anchored in the envelope of the virus, is the functional equivalent of TM. Hemagglutinin is a trimer that is composed of six subunits, three HA1 and three HA2.
In contrast to the cellular receptors recognized by retroviral Env proteins, all of which appear to be specific proteins (or glycoproteins), hemagglutinin binds with high specificity but low affinity to sialic acid residues that are found on most glycoproteins and glycolipids. The binding site for sialic acid is a pocket formed of several folds of the polypeptide chain of HA1 at its farthest point from the cell membrane. The ubiquitous distribution of sialic acid on cell surface molecules helps to explain the broad host range of influenza virus. The host range of retroviruses, on the other hand, is much more restricted, as would be predicted from their more specialized cellular receptors.
It is clear from a variety of studies that the mere binding of the viral glycoprotein and cellular receptor is not sufficient to cause fusion of the viral and cellular membranes. In both the influenza and retroviral systems, additional steps are involved. In the influenza system, a pH-dependent conformational change occurs in the hemagglutinin protein. This pH change is a direct result of the mechanism that the influenza virus uses to enter the cell (via endocytosis) and the subsequent drop in the pH inside the endosome when it matures. It should be emphasized here that this differs from what occurs in the life cycle of most retroviruses. Retroviruses rarely enter cells by endocytosis, and the fusogenicity of most retroviral Env proteins is not usually dependent on low pH. Instead, a high-affinity interaction of SU with the receptor acts in a more direct way to induce the conformational change.
How does the change in pH bring about influenza-virus-induced membrane fusion? In the structure of hemagglutinin (Fig. 8A), the HA2 protein contains a long carboxy-terminal α-helix (red) that is connected, via a loop, to a shorter amino-terminal α-helix (yellow). These two helices lie near each other in a hairpin-like structure. At the amino-terminal end of HA2 just before the second α-helix is a hydrophobic sequence thought to have the direct role of membrane insertion in the fusion process. In the inactive, high pH state, this fusogenic peptide is located near to the viral membrane. On exposure to low pH, conformational changes occur, and it has been postulated that the hairpin straightens out as the extended protein chain between the helices becomes part of a single long α-helix (Fig. 8B) (Carr and Kim 1993; Bullough et al. 1994), which in concert with the other HA2 molecules in the trimer forms a triple helix coiled-coil structure (Fig. 8C). This rearrangement would physically move the fusogenic peptide into a position where it could interact with the target cell membrane and initiate the fusion process (represented schematically in Fig. 9). The precise mechanism by which the fusogenic peptide mediates membrane fusion is still unclear, but it is possible to imagine that having one end of the HA2 protein embedded in the viral membrane and the other end embedded in the cellular membrane helps to bring the two membranes close enough together to initiate the process.
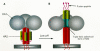
Figure 9
Schematic representation of the structural changes in the hemagglutinin trimer following acidification. (A) Neutral pH (virion-associated) form. (B) Acid pH (endosome-activated) form.
Why should this process be so complex? Why is this elaborate spring-loaded mechanism necessary? Presumably, the virus needs to distinguish, during its encounters with membranes, only those membranes that involve the surface of a susceptible cell. Moreover, the manufacture and transport of the viral protein and its subsequent display on the surface of the cell should do as little damage to the cell as possible.
To what extent does the influenza model describe the retroviral case? The analogy is tempting, particularly since there is, on the TM protein of retroviruses, a fusogenic peptide known to be necessary for viral entry. By inference, this peptide is thought to be buried within the Env oligomeric structure prior to activation and exposure through a conformational change. In addition, a region analogous to the extended “hairpin loop” of HA2 has been identified within the TM of the HIV Env protein and shown to have the capacity to both form a similar helical, coiled-coil structure and play a key part in viral fusion (Dubay et al. 1992; Wild et al. 1992, 1994; Chen et al. 1993). Furthermore, the recently solved three-dimensional structure of a 55-amino-acid fragment of the MLV TM protein that encompasses this region (Fig. 10A) shows that this retroviral component shares structural elements with the acidified HA2 triple-stranded coiled-coil (Fass et al. 1996). In contrast, the structure of a fragment of MLV SU that includes the receptor-binding region (Fig. 10B) is rather different from that of HA1 (Fass et al. 1997). This difference in structure most likely reflects the differences in the nature of the receptor as well as the differing roles of receptor interaction in entry of the two viruses. (This is discussed in more detail below under Membrane Fusion and Viral Entry.) At this point, however, some caution should be exercised. Although the influenza model is an exceptionally useful way to think about the interactions of retroviral Env proteins and their receptors, too little is known about the structure of retroviral Env proteins to make definitive statements about the similarities in the induction of membrane fusion during the initial stages of infection by influenza and retroviruses.

The ASLV Env Proteins
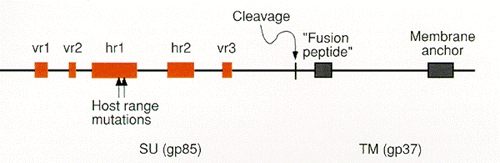
The ASLVs studied most thoroughly to date can be divided into five host-range subgroups, A through E (see above section on Receptores, ASLV; for review, see Vogt and Hu 1977). The observation that viral interference could be induced by soluble SU protein, together with a genetic analysis of viruses of different subgroups, has shown that subgroup specificity is defined by gp85 (SU) (Tozawa et al. 1970; Joho et al. 1975; Wang et al. 1976; Coffin et al. 1978). In the past several years, the nucleotide sequences of the env gene region of several viral isolates have been determined, and the precise location within the nucleotide sequence of the region encoding the mature env gene products has been defined by sequence analysis of the SU and TM proteins (Hunter et al. 1983).
By correlating differences in the receptor specificities of the env gene products of several ASLVs with amino acid sequence variation within the SU proteins, it has been possible to identify regions of the glycoprotein that contribute to receptor recognition. This type of comparison of the env genes of ASLV subgroups A–E revealed five discrete variable regions present in a highly conserved background (Schwartz et al. 1983; Dorner et al. 1985; Kan et al. 1985; Bova et al. 1986, 1988). The two larger variable regions are designated hr1 and hr2 (44–52 and 27–30 amino acids, respectively), and the three smaller regions are designated vr1, vr2, and vr3 (6–12 amino acids long; Fig. 11). The contribution that these variable sequences make to receptor binding has been assessed by constructing glycoproteins with different combinations of the variable domains and by analyzing the subgroup specificity of viruses containing them.
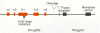
vr1 and vr2 sequences do not appear to be critical for receptor specificity. The host range of a B subgroup virus is not affected by replacement of its vr1 segment with a subgroup C sequence even though these vr1 domains show the greatest divergence (Dorner and Coffin 1986; Bova et al. 1988). Moreover, since variation at vr2 is observed only within subgroup B isolates, it is unlikely to have a role in defining to which receptor the virus binds.
The hr1, hr2, and vr3 sequences, on the other hand, are involved in receptor recognition. The NTRE-4 virus, which is a recombinant between Rous sarcoma virus (RSV)-Pr-B and an endogenous subgroup E virus related to RAV-0, encodes a chimeric env gene in which the B subgroup hr2 sequence has been replaced by that of the E subgroup gene (Dorner et al. 1985). The substituted hr2 confers an expanded host range on this virus which retains the capacity to utilize the B subgroup receptor but which can also utilize the subgroup E receptor on turkey cells (Tsichlis et al. 1980; Dorner et al. 1985).
Substitution of the subgroup E vr3 sequence into NTRE-4 (so that both hr2 and vr3 are from RAV-0) abolishes its ability to bind the B subgroup receptor and subtly modifies the subgroup E receptor-binding site so that it can bind the subgroup E receptor on chicken cells but not the subgroup E receptor on turkey cells. Thus, it seems likely that recognition of the turkey receptor by NTRE-4 requires an interaction between the E subgroup hr2 sequence and other sequences in Env that are missing when the E subgroup vr3 sequence is substituted (see Dorner and Coffin 1986).
It is important to point out that the B and E subgroup receptors on chicken cells appear to be different alleles of the tv-b gene and therefore that the differences between E and B Env proteins in their binding sites may be subtle and more amenable to retention of activity in recombinants between the two. This idea is supported by the observation that it is possible to select a mutant of subgroup B ASLV with a similarly extended host range. In contrast to NTRE-4, the two amino acids altered in this mutant are found in hr1 (Fig. 11) (R. Taplitz and J. Coffin, in prep.). Taken together, the host range of these viruses indicates that hr1, hr2, and vr3 are involved in receptor recognition and that the extent to which each of these contributes to the binding site varies with different receptors. Recognition of different receptors is also not necessarily mutually exclusive but can be complementary.
In contrast to the results with NTRE-4, several recombinants, in which the hr1 and hr2 variable regions from viruses of different subgroups were combined, encoded glycoproteins that were biologically inactive and unable to confer infectivity on virions. These results support the concept that interactions between variable regions are involved in receptor recognition. However, a defect at the stage of folding, intracellular transport, or incorporation into virions cannot be ruled out (Dorner and Coffin 1986).
The tropism of subgroup D viruses for mammalian cells has been mapped to sequences in and around the hr1 variable domain. However, the ability to infect mammalian cells may reflect characteristics of the Env protein that are distinct from recognition or binding of the receptor. In one case, a correlation was observed between infectivity for mammalian cells and temperature sensitivity of the virus. Thus, the differentiating feature of subgroup D and subgroup B viruses (which utilize the same receptor) may be a structural instability in Env that is important for efficient fusion with mammalian cells (Bova-Hill et al. 1991).
Although these types of analyses of recombinant viruses point to a role for hr1, hr2, and vr3 in receptor recognition, the nature of the binding site remains elusive. It is likely that the highly conserved sequences flanking these regions adopt a three-dimensional structure which is conserved overall and that the variable regions define specific binding domains within this framework. It is also possible that the receptor-binding sites involve both conserved residues and variable residues that are not unique to a subgroup, as well as subgroup-specific amino acids. Given the dissimilarity of the sequence and, presumably, structure of the A and B receptors, it is likely that the env genes of these viruses have a capability of recognizing diverse molecules from within otherwise highly conserved structures. This ability—analogous to immunoglobulin genes—would allow a virus the evolutionary flexibility to adapt to hosts lacking the usual receptor.
Murine Type-C Viruses
Like ASLV, the MLVs can be divided into several subgroups based on their env genes. These subgroups differ in the receptors they recognize. Each group includes viruses able to interfere with one another (Rein 1982; Rein and Schultz 1984). As discussed above, four subgroups of naturally occurring MLVs have been identified based on their receptor use in mouse cells: ecotropic, amphotropic, polytropic (mink cell focus-forming virus), and xenotropic. A fifth subgroup, defined by the 10A1 virus, which has an expanded host range due to its ability to bind both the amphotropic and GALV receptors, is apparently derived from recombination between an infecting amphotropic virus and an endogenous MLV-like sequence (Lai et al. 1982; Rein and Schultz 1984; Ott et al. 1990).
Early studies with soluble forms of the MLV gp70 (SU) showed that receptor binding was a property of this protein (DeLarco and Todaro 1976; Fowler et al. 1977). The SU proteins of MLVs contain a proline-rich segment, which is highly variable in sequence among different isolates and which separates a variable amino-terminal region from a highly conserved carboxy-terminal domain (Koch et al. 1983, 1984). As with the ASLVs, it has been possible to correlate differences in host range (receptor specificity) with sequence differences within the SU-encoding region of the env gene. A comparison of SU sequences from MLVs indicates that the amphotropic, 10A1, endogenous polytropic, and xenotropic sequences are closely related, with 64% identity among the amino acid positions (Stoye and Coffin 1987; Ott et al. 1990). Most of the amino acid differences among these nonecotropic viruses are clustered in three regions (see Fig. 7A). About 50 amino acids from the amino terminus is a region of variable length (VRA, 36–42 amino acids) that includes two conserved cysteine residues, but some amino acid variation. A second region of variable length (VRB, 22–29 amino acids) that includes a single conserved cysteine residue is found about 40 amino acids downstream from the first. The third variable region lies in the proline-rich domain and is about 50 amino acids long. The env sequences of ecotropic viruses are less closely related to those of the other four subgroups and also are quite variable relative to one another (Koch et al. 1983; Ott et al. 1990). Compared to their nonecotropic counterparts, the ecotropic sequences have a large insertion in VRA, a deletion in VRB, and numerous differences within the proline-rich region (see Fig. 7A) (Ott et al. 1990; Battini et al. 1992).
The amino-terminal region of the ecotropic gp70, which includes the first two variable sequences, determines its specificity for the ecotropic receptor. A fragment of gp70 from the ecotropic Fr-MLV that contains the amino-terminal 245 amino acids (VRA and VRB), but lacks the proline-rich region, retains the ability to interfere specifically with infection by ecotropic viruses both in a membrane-anchored form and as a secreted fragment (Heard and Danos 1991). The level of interference induced by expression of the secreted fragment was as great as that induced by a soluble form of SU and the intact ecotropic glycoprotein (Battini et al. 1994). Consistent with this result, deletion or insertional mutagenesis of the VRA/VRB region disrupted receptor recognition (Heard and Danos 1991; Gray and Roth 1993).
Replacement of the amino-terminal domain and the proline-rich region in an ecotropic virus, through recombination with an endogenous retroviral sequence, generates a virus whose Env glycoprotein binds the polytropic receptor rather than the ecotropic receptor (Bosselman et al. 1982; Vogt et al. 1986). Similarly, a chimeric Env protein in which the amino-terminal 240 amino acids (VRA and VRB) of the mature Mo-MLV SU protein are replaced by 159 amino acids (containing VRA and VRB) from the amphotropic (4070A) virus confers amphotropic host range (Morgan et al. 1993). Thus, the VRA region of SU has a predominant role in receptor choice by the ecotropic MLVs.
The contribution of different regions of nonecotropic gp70s to their receptor specificity has also been examined by construction of chimeric gp70 molecules. The amino-terminal (221-amino-acid) region of an amphotropic gp70 is sufficient to confer specificity for the amphotropic receptor on chimeric proteins containing polytropic or xenotropic downstream sequences that include the proline-rich segment (Battini et al. 1992; Ott and Rein 1992). In this case, however, both VRA and VRB are required for amphotropic receptor binding. Similarly, the VRA and VRB regions of both the polytropic (MCF 247) and xenotropic glycoproteins appear to interact to generate a functional receptor-binding domain. Although it is the VRA domain that defines whether the polytropic or xenotropic receptor can be recognized (Battini et al. 1992), the proline-rich hinge region of these viral glycoproteins also appears to be involved in the three-dimensional structure that binds the receptor (Ott and Rein 1992; Battini et al. 1994).
In most MLVs studied, then, the amino-terminal domain of gp70 determines receptor specificity, although the specificity can be modulated by interactions with downstream sequences. The significance of the variability in the proline-rich region is not clear. The positions of the prolines vary but represent about one third of the residues in this region. A dominant effect of the prolines in determining the structure within this region may allow greater variation at other positions, without affecting protein function.
Other Mammalian C-type Viruses
Other mammalian C-type Env proteins are quite closely related to those of the MLVs. Indeed, The FeLV-A SU protein is more similar to endogenous (ecotropic and xenotropic) MLV than endogenous and exogenous MLVs are to one another. The amino-terminal regions of FeLV SU proteins contain conserved regions closely related in sequence to those of MLVs and contain variable regions at similar positions (Elder and Mullins 1983; Riedel et al. 1986; Donahue et al. 1988). FeLV-B is most distinct from FeLV-A in this amino-terminal region, where there are four variable domains (vr1– vr4) (Stewart et al. 1986; Kumar et al. 1989; Neil et al. 1991; Sheets et al. 1992).
The GALV SU sequence is less closely related to that of MLV but retains similarity in the constant regions. All 16 of the cysteine residues conserved among the SU proteins of MLV are also present in the GALV and FeLV proteins, consistent with the idea that they have similar tertiary structures. Although env sequences of different FeLV subgroups differ within the variable regions, the FeLV-B and GALV proteins show little similarity in these variable amino-terminal domains (Battini et al. 1992) despite using the same receptor (Johann et al. 1992; Takeuchi et al. 1992). How then do the variable regions of these proteins contribute to receptor binding? It is possible that GALV and FeLV-B bind the same receptor through distinct molecular interactions; alternatively, the divergent sequences may fold into a common structure as is seen with the retroviral proteases (Skalka 1989).
Several observations have yet to be accounted for on mammalian retroviral infection of cells. First, not all viral isolates placed within a subgroup have the same host range on heterologous cells (Cloyd et al. 1985). Second, certain recombinant viruses have the host range of one subgroup but the interference properties of another subgroup (Cloyd and Chattopadhyay 1986). Third, sequence comparisons suggest that the known ecotropic viruses may themselves be recombinants, implying that the original ecotropic parent has not been identified (Stoye and Coffin 1987). Fourth, and most perplexing, is the observation that the patterns of interference seen in NIH-3T3 cells are altered in other cells, for example, the SC-1 cell line derived from a wild mouse or a cell line derived from M. dunni (Chesebro and Wehrly 1985). An understanding of these and other phenomena related to host range must await detailed understanding of the effect of env sequence heterogeneity on receptor interaction and the isolation of what is likely to be a very polymorphic set of viral receptors from the host cell.
HIV and Related Lentiviruses
Because of their direct relevance to a fatal human disease, HIV and the related primate lentiviruses have been the focus of a large body of recent work aimed at understanding the site and molecular interactions involved in receptor binding. This group of retroviruses presents quite a different situation, with regard to Env variation and receptor choice, from that described above for ASLVs and MLVs. With the primate lentiviruses, extensive variation is observed in the SU domain of different isolates and types, but a common primary receptor, CD4, is utilized by all. In this case, then, the conserved regions of the SU protein, rather than those that vary between strains, must combine to define the CD4-binding site.
Genetic and Immunological Mapping of the Receptor-binding Domain
As described above, a comparison of HIV-1 isolates led to the identification of five variable regions and four constant regions in gp120, designated V1–V5 and C1–C4 (see Fig. 6A) (Starcich et al. 1986; Willey et al. 1986; Modrow et al. 1987). Despite the considerable variation in primary sequence, all naturally occurring gp120 molecules retain the ability to bind CD4. Characterization of the interaction of gp120 with CD4 supports the concept that conserved residues in gp120 are involved in this process and also indicates that discontinuous segments of the linear SU protein sequence contribute to the binding site. Initially, a segment of the fourth conserved domain (C4) was implicated in CD4 binding, since monoclonal antibodies to gp120 that inhibit gp120-CD4 binding bind unique peptides from this region, and deletion of small portions of C4 from gp120, resulting in loss of the epitope recognized by the monoclonal antibody, also disrupt CD4 binding (Lasky et al. 1987; Cordonnier et al. 1989a,b; Sun et al. 1989). However, an analysis of the effects of insertion mutations and single-amino-acid changes throughout gp120 has indicated that in addition to those in the C4 domain, C2 and C3 residues also influence the CD4-binding ability of gp120 (see Fig. 6B) (Kowalski et al. 1987; Olshevsky et al. 1990). The CD4 binding of secreted gp120 molecules is not affected by deletion of the amino-terminal 62 residues or carboxy-terminal 20 residues, but deletion of additional residues from the termini disrupts CD4 binding, probably by preventing folding of gp120 into a functional molecule (Dowbenko et al. 1988; Pollard et al. 1992). The variable domains within SU do not have a direct role in receptor binding since a gp120 molecule lacking V1, V2, and V3 as well as the 62 amino-terminal and 20 carboxy-terminal residues still binds CD4 with an affinity similar to that of intact gp120 (Pollard et al. 1992; Wyatt et al. 1993). The binding of the CD4 receptor by gp120, then, involves the constant regions, and although much attention has been focused on the C4 region, it is but one region that contributes to the binding site; other conserved regions of gp120 that are separated in the sequence do so as well. HIV gp120 resembles in this regard both the ASLV SU and the HA1 protein of influenza virus in that different regions of the SU must come together in the three-dimensional structure to form the functional receptor-binding site.
Further understanding of the conformation of SU relative to CD4 binding comes from studies of the interaction of neutralizing and nonneutralizing antibodies with this molecule. Neutralizing antibodies for HIV-1 recognize both linear and discontinuous epitopes on gp120. Those that develop early in humans infected with the LAI strain of HIV-1 are directed primarily against linear neutralizing determinants within the V3 domain (Matsushita et al. 1988; Rusche et al. 1988; Javaherian et al. 1989). These antibodies do not block CD4 binding but appear to interfere with postreceptor-binding events that are involved in viral entry (Linsley et al. 1988; Skinner et al. 1988). Later in the course of HIV-1 infection in humans, antibodies with broader neutralizing capacity appear (Berkower et al. 1989; Robinson et al. 1990; Posner et al. 1991; Tilley et al. 1991). In essentially all HIV-infected individuals, these include antibodies that interfere with the binding of CD4 by gp120 (McDougal et al. 1986b; Schnittman et al. 1988; Kang et al. 1991). Most recognize discontinuous epitopes of gp120 only present on the native glycoprotein (Steimer et al. 1991; Haigwood et al. 1992; Moore and Ho 1993). Unlike the early antibodies directed against V3, which are highly strain-specific, such antibodies recognize gp120 molecules from a diverse range of HIV-1 isolates and can also neutralize cell-line-adapted strains (Robinson et al. 1990; Ho et al. 1991; Posner et al. 1991).
Antibodies can be used to map functionally important regions of the SU protein by testing their binding to specific mutant proteins. Such studies have revealed that epitopes recognized by four monoclonal antibodies involve amino acids in seven discontinuous conserved regions of gp120, four of which overlap regions previously shown to be important for CD4 binding (Thali et al. 1991b, 1992, 1993; McKeating et al. 1992). Thus, in the native gp120 structure, each of the conserved domains, C1–C4, is probably folded in such a way that a single antibody molecule can interact with residues from each, whereas the binding pocket for CD4 is formed from residues in C2–C4. Because CD4-blocking antibodies interact with residues outside, as well as within, the CD4-binding domain, it is possible for the virus to develop resistance to neutralization through mutations in the former sites while retaining an intact CD4-binding domain. An analogous situation is observed in poliovirus, where the receptor-binding site forms a “canyon” on the surface of the virus that is inaccessible to antibody and allows surface residue alterations to occur without changing the binding pocket itself (Rossmann 1989). Similarly, with the influenza virus neuraminidase, neutralizing epitopes involve amino acid residues that are located on the rim of the substrate-binding pocket, so that neutralization-escape mutations do not involve the highly conserved active site residues (Air et al. 1989).
Antibody binding can also be used to infer the structure of the native protein. Monoclonal antibodies to continuous epitopes in the variable V1, V2, and V3 domains are accessible in both the monomeric and oligomeric molecules (Moore et al. 1994). In contrast, most of the epitopes in conserved regions of gp120 are not well exposed on the surface of monomeric gp120; the few conserved continuous epitopes (amino- and carboxy-terminal regions) exposed on the monomer molecule are inaccessible on an oligomeric form of the glycoprotein expressed on the surface of infected cells. In a more extensive survey, a soluble form of CD4, together with 46 monoclonal antibodies able to bind native monomeric gp120, was used to generate a competition binding matrix. On the basis of these data, a model of the epitope distribution on the gp120 surface has been constructed, which predicts that the binding sites for CD4 and for neutralizing monoclonal antibodies are located on one face of the gp120 glycoprotein, consistent with this side of the molecule being exposed on an assembled oligomeric glycoprotein complex. A second face of gp120 that is presumed to be inaccessible on the assembled complex contains a number of epitopes for nonneutralizing antibodies (Moore and Sodroski 1996). Thus, it is likely that CD4 (and antibodies that compete for CD4 binding) recognizes scattered, discrete but conserved residues that come together in a tightly folded three-dimensional structure to form a high-affinity binding site.
The failure of antibodies against conserved Env sequences to recognize native protein implies that the protein is structured in such a way as to resist neutralization of virus by antibody in the course of infection in vivo (Moore and Ho 1995). Structural features that probably relate to this resistance include the abundance of glycosylation sites and the V regions that may hide functionally important conserved regions from antibody binding. In support of this idea is the observation that HIV isolates are initially resistant to neutralization by antibodies against conserved regions, but become sensitive after a few passages in cell culture where selection pressures are different (Moore and Ho 1995). This neutralization resistance property of the Env protein is likely to be important for continued replication of HIV in the presence of high concentrations of antibody in vivo (Chapter 11) and in the failure of vaccines based on free Env protein to provide significant protection against infection (Chapter 12).
Nonreceptor Determinants of Early Events in Infection
HIV Tropism and the V3 Loop
The construction of chimeric proviruses has allowed the mapping of macrophage tropism determinants to a region within the viral env gene that includes the V3 domain (for review, see Moore and Nara 1991). A 20-amino-acid sequence from the V3 region of one M-tropic isolate is sufficient to confer macrophage tropism on HTLV-IIIB, a T-cell-line-tropic isolate (Hwang et al. 1991). Moreover, certain single-amino-acid changes within the V3 loop can alter cell tropism of HIV-1 (Takeuchi et al. 1991; Ivanoff et al. 1992), and others can abrogate or greatly reduce cell fusion and viral infectivity without blocking gp120-CD4 binding (Freed et al. 1991; Bergeron et al. 1992; Grimaila et al. 1992; Ivanoff et al. 1992; Page et al. 1992). Similar effects on tropism and infectivity have been observed in mutagenic analyses of the V3-equivalent domains of SIV and HIV-2 (Freed and Myers 1992; Hirsch et al. 1994; Kirchhoff et al. 1994). The function of the V3 domain in viral fusion/entry has not been elucidated. It may interact directly with the recently identified second receptor for HIV. In addition, both genetic and immunochemical analyses suggest that the V3 loop and the C4 domain are topologically associated. Amino acid sequences within V3 can influence CD4 binding (Willey et al. 1994b), and amino acid changes in one region can influence antibody accessibility to the other (Wyatt et al. 1992; Moore et al. 1993c). Thus, it is possible that the V3 loop is topologically associated with the CD4-binding domain and defines whether interactions with CD4 molecules in particular cell membrane contexts are functional or otherwise.
Other Determinants of Tropism
Despite a clear role in defining monocyte-macrophage tropism, it would be wrong to infer that the V3 loop is the only region of the HIV glycoprotein that influences HIV-1 tropism. Adaptation of one M-tropic strain of HIV-1 to growth in a T-cell line accompanied by a broadened tropism for promonocytic cells can result from two amino acid changes in the C4 region of gp120 and the fusion peptide region at the amino terminus of gp41. The adapted variant virions show increased binding of sCD4, and although each mutation individually increases sCD4 association, both changes are required for maximal binding and infectivity (Willey et al. 1994a). Similarly, substitution of fusion peptide sequences from HTLV-1 and SIV into gp41 of HIV-1 does not abrogate viral infectivity but alters the spectrum of efficiencies with which the viruses replicate on a series of different T-cell lines (Bergeron et al. 1992). Finally, the ability of certain SIV isolates to infect specific human T-cell lines appears at least in part to depend on the density of glycoprotein spikes on the viral surface, suggesting that for certain target cells, a threshold of glycoprotein/receptor interactions is required for viral entry to proceed (Johnston et al. 1993; Zingler and Littman 1993).
The complexity of host-range restriction (tropism) observed with HIV and SIV on CD4-expressing cells is consistent with complex patterns of additional accessory molecules necessary to confer susceptibility to HIV infection. As described above, chemokine receptors serve as specific accessory membrane proteins to facilitate entry into macrophages and T-cell lines, but it is likely that a subtle combination of differences in both the viral glycoprotein and the CD4 receptor itself also influences tropism. In gp120, such differences could include the accessibility of the CD4-binding site, the surface density of the carbohydrate groups, and the ease with which conformational changes could occur. In CD4, they might reflect the context of the receptor in terms of other surface molecules, its surface density, and the fluidity/composition of the membrane.
HIV-2 and SIV isolates also use CD4 as their cell surface receptor, but they have cell surface requirements different from those of HIV-1. HIV-2 strains have been shown to infect a range of CD4-expressing cells including the human U87 glioma cell line and those of nonprimate origin (mink, cat, rabbit) which are nonpermissive for HIV-1 infection (see Table 5) (Clapham et al. 1991; Dragic and Alizon 1993). Similarly, SIV strains show a more restricted capacity for infection of human T-cell lines, but they can infect the U87 cell line as well as nonhuman primate cells expressing CD4 (Koenig et al. 1989; Clapham et al. 1991). At least some of the restriction SIV displays for human cell lines have been mapped genetically to the fusion peptide region of TM (Bergeron et al. 1992).
Publication Details
Copyright
Publisher
Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY)
NLM Citation
Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997. Retroviral Env Gene Products: Receptor Binding and Host-Range Determinants.