Genome editing in human pluripotent stem cells*
Authors
Derek T Peters,
1 Chad A Cowan,
1,2 and
Kiran Musunuru1,3.
*Affiliations
1 Department of Stem Cell and Regenerative Biology, Harvard University; Harvard Stem Cell Institute, Cambridge, Massachusetts 02138, USA
2 Center for Regenerative Medicine, Massachusetts General Hospital, Boston, Massachusetts 02114, USA
3 Division of Cardiovascular Medicine, Brigham and Women's Hospital, Boston, MA 02115, USA
* Kiran Musunuru, Harvard University, Sherman Fairchild Biochemistry Bldg 160, 7 Divinity Ave, Cambridge, MA 02138, USA, E-mail: kiranmusunuru@gmail.com / Phone: (617) 496-5361 / Fax (617) 496-8351
Published April 29, 2013.
Genome editing is used to make targeted modifications to the genome of eukaryotic cells. There are many potential applications of genome editing in human pluripotent stem cells (hPSCs) including the generation of knockout and reporter cell lines. This protocol describes a system for efficient genome editing in hPSCs using engineered transcription activator-like effector nucleases (TALENs) or clustered regularly interspaced short palindromic repeat (CRISPR) technology.
Introduction
Genome editing in mammalian cells is the process of generating genetically modified cell lines using engineered sequence-specific nucleases. Custom-engineered nucleases are introduced into cells, in which they create targeted double-strand breaks (DSB) at a genomic site of interest for the purpose of creating insertion/deletion mutations (indels) or facilitating homologous recombination. In the later approach, a short single-strand oligonucleotide (ssODN) or double-strand plasmid is used to introduce a desired DNA sequence into the genome. Transcription activator-like effector nucleases (TALENs) have been used successfully for genome editing in human pluripotent stem cells (hPSCs) (Hockemeyer et al., 2011), and custom TALEN pairs can be readily designed and constructed. Similarly, clustered regularly interspaced short palindromic repeat (CRISPR)/Cas systems have been demonstrated to achieve genome editing in hPSCs (Mali et al., 2013).
There are an array of potential applications for genome editing in hPSCs including the following: knocking out a gene of interest by inducing an indel mutation, knocking in a disease-associated DNA variant, generating reporter cell lines, and correcting a causal mutation in a patient-specific induced pluripotent stem cell (iPSC) line. Our laboratory has developed a system for efficient TALEN-mediated genome editing in hPSCs (Ding et al., 2013a) and used this system to create several cellular disease models. We have also adapted this system for CRISPR-mediated genome editing (Ding et al., 2013b).
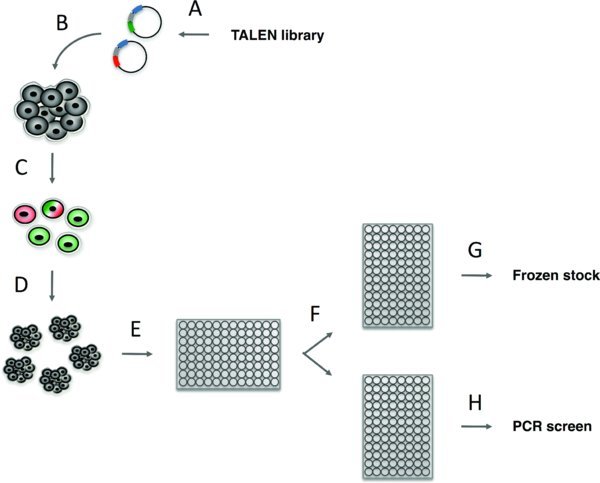
Flowchart
summarizes the steps necessary to accomplish genome editing in hPSCs using our system, beginning with the design and construction of custom TALENs (or CRISPRs) and ending with the identification and expansion of successfully modified hPSC lines. Section 3 describes the process of designing and constructing TALENs using our publicly available vector system (http://www.addgene.org/TALEN_genome_editing_collection). Section 4 describes the process of designing CRISPRs. Section 5 provides a detailed protocol for using TALENs or CRISPRs in hPSCs and identifying modified clones. Our system does not require the use of antibiotic selection to identify modified hPSC lines; for gene targeting with antibiotic selection, skip section 5.1.3 of the protocol.
Generating custom TALENs
Protocol
TALEN binding site criteria and TALEN design
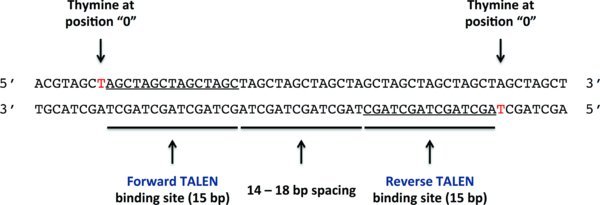
TALEN binding sites are chosen such that the forward and reverse TALENs will flank the desired genome-editing site. TALEN-induced DSBs, which are the substrate for the genome-editing process, occur in the region between the TALEN binding sites. Choose TALEN binding sites that satisfy the following criteria ():
Each TALEN binding site is 15 bp long
The spacing between the forward and reverse TALEN binding sites is between 13 and 13 bp; 16 bp is optimal
The forward TALEN binds in the 5′ to 3′ direction on the forward DNA strand
The reverse TALEN binds in the 5′ to 3′ direction on the reverse DNA strand
The position 0 nucleotide (the nucleotide just 5′ to the first position of the TALEN binding site) should ideally be a T for both TALENs
TALEN subcloning overview
Once TALEN binding sites are chosen, custom forward and reverse TALENs are constructed that are specific to the 15-bp binding sites, each in the 5′ to 3′ direction. Each TALEN consists of a sequence-specific 15mer DNA-binding domain as well as a nuclear localization signal (NLS), invariant N-term and C-term domains, and a FokI nuclease domain. For the purpose of subcloning, the 15mer DNA-binding domain is broken into four “positions”: three tetramer-recognition pieces (positions I, II, and III) and one trimer-recognition piece (position IV).
Our TALEN construction library comprises a collection of 832 plasmids containing all of the position I, II, III, and IV pieces necessary to generate a TALEN for any 15-bp target sequence (as well as two backbone vectors; see below). Using this library, a custom TALEN is generated through a five-piece subcloning ligation involving three sequence-specific tetramer-recognition pieces, one trimer-recognition piece, and an expression vector backbone (pTAL). There are two distinct pTAL vectors (pTAL_GFP and pTAL_RFP); the forward and reverse TALENs must be subcloned into different pTAL vectors so that the two FokI domains of the TALENs will correctly heterodimerize once they are bound to their target sites in the genome (the FokI domains are designed not to homodimerize). Begin by determining which position I, II, III, and IV plasmids you will use to generate sequence-specific TALENs for your chosen binding sites.
Digest tetramer and trimer pieces for TALEN subcloning
Digest and gel-purify the appropriate position I, II, III, and IV plasmids (e.g., for the binding site “GCCGGAGCTGTCGAC” digest the “I-GCCG,” “II-GAGC,” “III-TGTC,” and “IV-GAC” plasmids). Proceed as follows:
If starting with a glycerol stock or agar stab culture, inoculate into LB with ampicillin and/or kanamycin (use both antibiotics for the most stringent selection), grow, and miniprep
Setup digest: 5--15 uL plasmid DNA, 2 uL Tango buffer, 1 uL 20 mM DTT, 1 uL BsmBI enzyme, dH2O to 20 uL reaction volume; optional: can add 1 uL NcoI to the reaction (adjust dH2O so that final reaction volume is 20 uL) to digest the vector backbone and reduce carryover of uncut vector to subsequent steps
Incubate several hours at 37°C
Run digests on agarose gel; expected insert sizes are about 425 bp for positions I, II, and III and about 280 bp for position IV
Gel-purify each insert using the QIAEX II kit (highly recommended) and elute into 20 uL elution buffer (EB)
Digest pTAL backbone vectors for TALEN subcloning
Each of the pTAL plasmids contains the CAG promoter, all parts of the TALEN protein except the DNA-binding domain, and either green fluorescent protein (GFP; specifically EGFP) or red fluorescent protein (RFP; specifically mCherry). Upon ligation of the tetramer/trimer pieces, the plasmid will co-express (via a viral 2A motif) the TALEN and the fluorescent protein. Each pTAL plasmid should be digested and gel-purified (can be done in parallel with the tetramer/trimer plasmids, or can be done beforehand to make stocks for use in numerous TALEN ligations). Proceed as follows:
If starting with a glycerol stock or agar stab culture, inoculate into LB with ampicillin, grow, and miniprep
Setup digest: 5 uL plasmid DNA, 2 uL Tango buffer, 1 uL 20 mM DTT, 2 uL BmsBI enzyme, 10 uL dH2O to 20 uL reaction volume
Incubate several hours at 37°C
Heat inactivate at 75°C for 10 minutes, then cool to room temperature
Add 27 uL dH2O, 3 uL New England Biolabs (NEB) buffer III, and 1.5 uL CIP to make a 50 uL reaction volume
Incubate at least 30 min at 37°C
Add 1 uL 0.5 M EDTA
Heat inactivate at 75 degrees for 10 minutes, then cool to room temperature
Add 50 uL dH2O and mix; final reaction volume should be 100 uL
Add 100 uL phenol/chloroform/isoamyl alcohol and vortex vigorously (be careful not to let contents leak out of tube)
Spin at maximum speed in microcentrifuge for 5 minutes
Carefully remove top (aqueous) phase 20 uL at a time, avoiding the white interface, and transfer to another tube; do this 4 times for total of 80 uL
Run 80 uL on an agarose gel (use wide, deep wells); expected size is about 6.5 kb
Gel-purify the vector (there should be no discernible insert) using the QIAEX II kit, elute into 20 uL elution buffer (EB)
TALEN subcloning
Assemble gel-purified tetramer and trimer inserts and pTAL vector via a five-piece ligation:
Set up ligation: 0.875 uL position I insert DNA, 0.875 uL position II insert DNA, 0.875 uL position III insert DNA, 0.875 uL position IV insert DNA, 0.5 uL pTAL vector DNA, 0.5 uL 10× T4 ligase buffer, 0.5 uL T4 ligase to make a final 5 uL reaction volume
Mix and incubate for several hours at room temperature
Transform into competent bacteria (e.g., DH10B)
Plate onto LB agar plate with ampicillin and grow colonies at 37°C
Screen colonies (putative TALEN constructs) for appropriate inserts:
Pick colonies into 5 mL of LB-ampicillin (typically 5--10 colonies are picked from each plate, i.e., for each TALEN)
Miniprep cultures, elute in 35--50 uL elution buffer
Digest 2 uL miniprep DNA with XbaI and BamHI for at least 30 min (the digestion may be somewhat inefficient and not go to completion in that time frame)
Run digests on agarose gel
The correct insert size is about 2.1 kb; may see evidence of incomplete plasmid digest
Sequence plasmids with correctly sized inserts using the primers TALseq+ (5′-GGCCAGTTGCTGAAGATCG-3′) and TALseq– (5′-CGCTACAAGATGATCATTAGTG-3′), which will read from either end of the insert and should cover the full DNA-binding domain if using a high-quality commercial DNA sequencing service (need at least 850 bp of reliable sequence from each read). Merge the DNA sequences via the overlap at the ends of the sequence reads, “translate” into amino acid sequences, and check to insure that the “code” of the DNA-binding domain is correct ().
Maxipreps of TALEN plasmids
Once the forward and reverse TALENs are sequence-confirmed, produce maxiprep quantities of endotoxin-free TALEN plasmid DNA in preparation for delivery of the TALENs to hPSCs via electroporation. Grow maxiprep cultures in LB-ampicillin. Elute or resuspend (depending on maxiprep protocol) plasmid DNA in dH2O to concentrations between 1 ug/uL and 3 ug/uL. 15 to 25 ug of each TALEN plasmid will be needed for each electroporation (see Section 5 for details).
Designing a knock-in ssODN
If an ssODN will be used to introduce a desired sequence into the genome, design a 60--70 bp oligonucleotide (matching the antisense strand if targeting a coding region; otherwise, the oligonucleotide can match either strand) with the non-homologous nucleotide(s) located near the center of the oligonucleotide sequence. To aid in screening for hPSC clones that correctly incorporated the desired sequence, it is helpful if the ssODN is designed to insert or remove a restriction site. To insert a restriction site into a coding sequence, consider incorporating a synonymous nucleotide substitution that creates a new restriction site without changing the protein-coding sequence into the ssODN.
Obtain a salt-free oligonucleotide produced at an appropriate scale (e.g., 50 nmol); 30 ug of the oligonucleotide will be needed for each electroporation (see Section 5 for details). PAGE purification is not necessary for oligonucleotides less than 70 bp long. Resuspend the oligonucleotide in dH2O to a concentration of 2 ug/uL.
Materials
TALEN construction library (available from Addgene)
LB, LB agar, ampicillin, kanamycin (any source)
BsmBI enzyme (Thermo Scientific/Fermentas)
Tango buffer (included with BsmBI enzyme)
20 mM DTT (any source)
dH2O (any source)
NcoI, XbaI, and BamHI enzymes (New England Biolabs or other source)
QIAEX II kit (Qiagen)
NEB buffer III (New England Biolabs)
CIP (New England Biolabs or other source)
0.5 M EDTA (any source)
Phenol/chloroform/isoamyl alcohol (any source)
T4 ligase (New England Biolabs or other source)
T4 ligase buffer (included with T4 ligase)
Competent bacteria cells (e.g., DH10B), either homegrown or commercial
TALseq+/TALseq– primers and ssODN (any source)
Troubleshooting
TALEN assembly ligations
If the TALEN assembly ligation repeatedly fails to produce any colonies or if none of the screened colonies have an insert that is the correct size, consider repeating the tetramer/trimer vector and/or backbone vector digests and gel purifications before repeating the assembly ligation.
If the TALEN ligation products have inserts that are ∼300--400 bp in size, these likely represent uncut or partially cut tetramer/trimer vector DNA that has been propagated through the steps of the protocol and ended up in the final ligation reaction. Adding NcoI to the original tetramer/trimer vector digestions can minimize this “background” vector.
If the TALEN ligation products have inserts that are the correct size but sequencing shows that the final 15mer DNA-binding domain is incorrect, consider sequence-confirming the tetramer/trimer plasmids (sequence with the universal T7 primer; expected sequences are available on the Addgene page) that were used in Section 3.1.3.
Sequence-confirming assembled TALENs
Sequencing the final TALEN construct may be difficult due to the reparative TAL repeat domain sequences; high-quality, concentrated plasmid DNA (grown from >5 ml culture and eluted in a small volume, e.g., 35 uL) is best for sequencing.
Maxipreps of TALEN plasmids
TALEN plasmids are relatively unstable; when doing maxipreps, use LB broth with fresh ampicillin and do not grow the maxiprep culture for more than 18 hours. Consider inoculating the maxiprep culture from a medium sized LB-ampicillin culture (<50 mL) grown for a few hours. In addition to using a spectrophotometer to quantitate the maxiprep DNA, digest a small amount of DNA and run it on an agarose gel to confirm that the actual amount of digested DNA matches the expected amount.
Generating custom CRISPRs
Protocol
Cas9/CRISPR binding site criteria and CRISPR design
Cas9/CRISPR binding sites are chosen to conform to the sequence G(N)19NGG; the CRISPR RNA component hybridizes with the first 20 DNA nucleotides, and the Cas9 protein recognizes the NGG motif. CRISPR-induced DSBs occur three nucleotides proximal to the NGG motif. The Cas9/CRISPR can recognize and bind to the motif on either DNA strand. Choose Cas9/CRISPR binding sites that satisfy the following criteria:
Match the G(N)19NGG sequence in proximity to the desired target site; usually, but not always, there will be a matching sequence within 16 nucleotides of the target site
Ideally, the binding site should not exactly match or mismatch by one nucleotide any other sequence in the genome
Cas9/CRISPR overview
The Cas9 component is invariant. Cas9 is expressed from the pCas9_GFP plasmid, which is identical to the pTAL_GFP plasmid except that the TALEN has been replaced with a human-codon-optimized Cas9 gene. This plasmid co-expresses Cas9 and GFP.
The CRISPR RNA component is expressed as a small guide RNA (gRNA) from the human U6 promoter. The 5′ end of the gRNA contains the 20-bp sequence [G(N)19]that hybridizes to the DNA target site; accordingly, changing this 20-bp sequence is all that is necessary to direct the complex to a different site in the genome. Any of a variety of methods can be used to specify the 20-bp sequence. The gRNA Empty Vector from George Church's laboratory (Mali et al., 2013) can be obtained from Addgene (https://www.addgene.org/CRISPR/Church/); protocols for inserting the 20-bp sequence into this vector is available there (https://www.addgene.org/static/cms/files/hCRISPR_gRNA_Synthesis.pdf).
Maxipreps of Cas9/CRISPR plasmids
Once the gRNA plasmid is sequence-confirmed, produce maxiprep quantities of endotoxin-free Cas9 and gRNA plasmid DNA in preparation for delivery of the Cas9/CRISPR components to hPSCs via electroporation. Grow maxiprep culture of pCas9_GFP in LB-ampicillin; if using a gRNA plasmid based on the Church laboratory's vector, grow maxiprep culture in LB-ampicillin/kanamycin. Elute or resuspend (depending on maxiprep protocol) plasmid DNA in dH2O to concentrations between 1 ug/uL and 3 ug/uL. 15 to 25 ug of each plasmid will be needed for each electroporation (see Section 5 for details).
Designing a knock-in ssODN
Same as for TALENs (see Section 3.1.7).
Genome editing in hPSCs
Protocol
Expansion of hPSCs and preparation of conditioned media
Expand an hPSC line (e.g., HUES 9, Cowan et al.,
2004) to 15-cm dishes. One 15-cm dish is used per electroporation; typically 1 to 4 electroporations are performed for a particular genome-editing experiment. Culture the cells using feeder-free adherent culture conditions in chemically defined mTeSR1 media supplemented with penicillin/streptomycin on dishes pre-coated with Geltrex matrix (250 uL Geltrex per 15-cm dish) (Schinzel et al.,
2011). Passage cells with Accutase [diluted 1:3 with calcium- and magnesium-free PBS (PBS–/–)] dissociation.
Replace mTeSR1 with fresh media (15--20 mL) every 24 hours; collect conditioned mTeSR1 (except during first media change, 24 hours after plating) until the cells are approximately 80% confluent, replace with fresh mTeSR1.
Make “post-FACS recovery media” using conditioned mTeSR1: 1 volume conditioned mTeSR1, 1 volume fresh mTeSR1, 35 uL 10,000× bFGF per 500 mL total media volume; filter-sterilize using tissue-culture media filter bottle (note: gentamicin can be added to the media for a few days following FACS to prevent FACS-related bacterial contamination).
TALEN or Cas9/CRISPR delivery by electroporation
Electroporate cells once they have grown to approximately 80% confluence:
Pre-treat cells with 10 uM ROCK inhibitor (Y-27632) for 3--4 hours prior to electroporation; add ROCK inhibitor directly to media.
Dissociate cells with Accutase; assure single cell dissociation.
Count cells and resuspend 10-12×10
6 cells (roughly the number of cells on a 15-cm dish) in 800 uL ice-cold PBS–/– containing 50 ug of the TALEN pair (25 ug of each plasmid) or a mix of 30 ug of the TALEN pair (15 ug of each plasmid) plus 30 ug ssODN or double-strand plasmid DNA for each electroporation; alternatively, setup one electroporation per 15-cm dish of cells without counting the cells. Perform 1-4 electroporations simultaneously. If using the Cas9/CRISPR system, resuspend the cells in 800 uL ice-cold PBS–/– containing 25 ug pCas9_GFP plasmid and 25 ug gRNA expression plasmid or 15 ug pCas9_GFP plasmid, 15 ug gRNA plasmid, and 30 ug ssODN or double-strand plasmid DNA for each electroporation. Note: it may be useful to also setup a “mock electroporation”; see
Section 5.1.3, step 6. The mock electroporation can be smaller in scale: use one-third the number of cells.
Transfer the 800 uL cell/DNA mixture into a 0.4 cm electroporation cuvette and incubate on ice for 5 minutes. If also performing a mock electroporation, transfer the 800 uL cell/PBS mixture (no DNA added) into a separate electroporation cuvette.
Electroporate using the following settings: 250 V and 500 uF (time constant should be between 10 and 14 seconds).
Quickly add 0.5 mL of mTeSR1 to each cuvette and transfer the contents of the cuvettes into 15-mL centrifuge tubes containing 2 mL of mTeSR1 per electroporation.
Gently pellet cells (200 g, 5 min, 25°C).
Gently resuspend the pellet in 5--10 mL mTeSR1.
Plate cells to one Geltrex-coated 15-cm dish per cuvette for a total volume of 20 mL mTeSR1 per dish with 10 uM ROCK inhibitor. If also performing a mock electroporation, plate cells to one Geltrex-coated 10-cm dish (100 uL Geltrex per 10-cm dish) for a total volume of 10 mL mTeSR1 with 10 uM ROCK inhibitor. Note: if performing genome editing with antibiotic selection, the post-electroporation plating density should be optimized so that isolated antibiotic resistant hPSC clones are generated over the course of antibiotic selection.
The day after electroporation, replace media with fresh mTeSR1 without ROCK inhibitor. Note: if using antibiotic selection, begin selection 24 to 48 hours after electroporation and continue selection until suitably sized antibiotic-resistant colonies appear (see
Section 5.1.4).
Fluorescence-activated cell sorting (FACS) enrichment and clonal isolation
The following steps are used for efficient genome editing without antibiotic selection; proceed to Section 5.1.4 if using selection.
After ∼48 hours following electroporation, confirm successful delivery of TALEN plasmids (or Cas9/CRISPR plasmids) by visualizing some green and/or red fluorescent marker expression using fluorescence microscopy (optional); proceed with FACS enrichment of fluorescence-expressing cells.
Dissociate cells with Accutase; assure dissociation to single cells. Combine cells from multiple replicate electroporations into a single tube.
Carefully resuspend cells in about 500 uL PBS–/– with 10 uM ROCK inhibitor, pass through a 35-um cell strainer cap into a FACS tube, and dilute with additional PBS–/– as needed. Higher cell concentrations will allow for more rapid FACS and possibly greater cell viability (see
Section 5.3.3) but may increase the risk of cell clumping during FACS.
Prepare 1.5 mL Eppendorf tube (for collection of cells) containing 700 uL mTeSR1 with 10 uM ROCK inhibitor.
Keep cells and collection tube on ice.
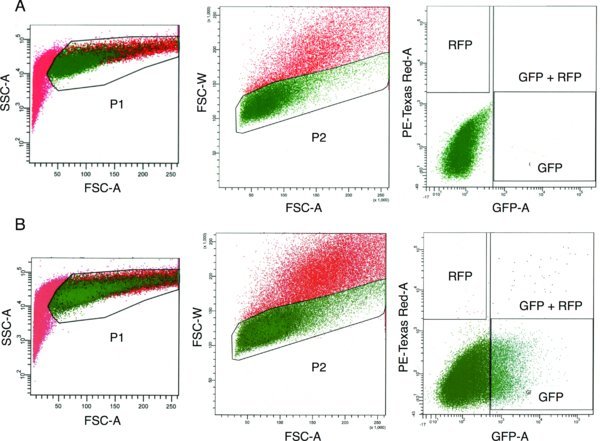
Using FACS, collect single cells that express green and/or red fluorescence into Eppendorf tube containing mTeSR1 (). Use unelectroporated or mock-electroporated (PBS only) cells to set gating for fluorescence ().
Plate post-FACS cells on Geltrex-coated 10-cm dishes at a density of 15,000 cells per 10-cm dish in post-FACS recovery media with 10 uM ROCK inhibitor. Plating density may be increased up to 50,000 cells per 10-cm dish if too few colonies are obtained (see
Section 5.3.3).
Change media with fresh post-FACS recovery media (without ROCK inhibitor) every 24 hours. Once multi-cellular colonies are clearly visible (typically 2 or 3 days after FACS), mTeSR1 can be used instead of recovery media.
Clone expansion and freezing
Begin picking colonies (typically 12 to 14 days after electroporation) when they are about the size of a nickel under the microscope with a 10× objective (A); do not allow colonies to grow too large and merge.
Coat a 96-well plate with Geltrex, using the same amount of Geltrex as for a 10-cm plate (100 uL Geltrex per plate).
Wash the colony plate with calcium- and magnesium-free PBS (PBS–/–).
Add 12 mL fresh PBS–/– to the plate and return the plate to the incubator for about 5 minutes to allow the cells to begin to detach (this greatly facilitates re-plating into the 96-well plate).
Prepare the 96-well plate with 120 uL mTeSR1 per well plus 10 uM ROCK inhibitor (not including volume of PBS/cells that you will be transferring into the wells).
Using a microscope setup in a tissue-culture hood or colony-picking hood pick colonies using a P200 pipette (set to about 45 uL) with filter tips by scraping the colony into small clumps of cells, collecting the cells with the pipette, and gently pipetting cells into a well of the 96-well plate. Use a new pipette tip for each colony. Try to collect all of the dissociated cells from a scraped colony; excess dissociated cells will float away in the PBS and may be accidently collected when subsequent colonies are picked. Pick colonies until a suitable number of colonies are isolated or the 10-cm plate contains a significant number of unwanted floating cells that cannot be avoided (typically after about 1 hour of picking). Note: the efficiency of TALEN-mediated genome editing using this system typically results in the generation of modified hPSC lines at a rate of 5% to 20% among the total number of colonies picked; Cas9/CRISPR-mediated genome editing appears to be even more efficient.
Change the medium (120 uL per well) in the 96-well plate every 24 hours (optional: add gentamicin to the media for a few days to prevent picking-related bacterial contamination) until the wells are about 80% confluent (typically 4 days after picking; ), when they can be split and frozen. Refer to
Section 5.3.5 if the cell density varies significantly from well to well on an individual 96-well plate.
Coat 1 or 2 Geltrex-coated 96-well plates per 96-well plate being passaged; 1 plate is for making DNA and 1 plate is for passaging and serves as an optional backup plate.
Prepare the Geltrex-coated 96-well plate(s) with 133 uL of mTeSR1 plus 10 uM ROCK inhibitor per well (the amount of ROCK inhibitor added should be based on the final volume per well, which is 200 uL; see step number 13).
Prepare an uncoated 96-well plate, which will be used for freezing.
Aspirate media from the original 96-well plate; wash once with 150 uL PBS–/– per well and aspirate.
Add 35 uL Accutase (1:3 diluted in PBS–/–) per well; treat in incubator for 5--10 min until most of the cells are detached from the plate (lightly tapping the side of the plate near the end of the treatment can help detach the cells).
Add 165 uL mTeSR1 per well directly to the plate.
Using a multichannel pipette, distribute 66 uL of cells (one third of the total volume of cells per well) to each of the 2 or 3 96-well plates—DNA plate, backup plate (optional), and freezing plate—and pipette up and down gently 1.5 times before disbursing the cells to ensure that the cells are equally distributed. Return the cells to the incubator.
Add 70 uL mFreSR per well to the freezing plate; wrap plate with Parafilm and move to a Styrofoam container at –80°C for freezing and storage.
On the next day, exchange the media on the new 96-well plates with 120 uL fresh mTeSR1 per well.
Change media every 24 or 48 hours.
Clone screening
Isolate genomic DNA (gDNA) from the cells in the 96-well plate designated for making DNA when the cells in each well are about 90% confluent:
Wash twice with 150 uL PBS per well.
Add 50 ul 96-well lysis buffer per well: 10 mM Tris pH 7.5 (or 8.0), 10 mM EDTA, 10 mM NaCl, 0.5% sarcosyl, 40 ug/mL proteinase K (added fresh before using the buffer).
Cover the plate with PCR plate film, replace the plate top, wrap in Parafilm, and wrap in plastic wrap or place in sealed plastic box with damp paper towel.
Incubate at 56--60°C overnight or for ≥6 hours.
Precipitate DNA by adding 100 uL cold 95% ethanol (not 100%) plus 75 mM NaCl (150 uL 5 M NaCl per 10 mL ethanol) per well; leave at –20°C for 2 hours.
Spin plate for 5 minutes at 1000 rpm.
Carefully decant liquid out of plate; the gDNA should stick to the bottom or side of each well.
Wash three times with 150 uL 70% ethanol per well; carefully decant after each wash.
Let residual ethanol evaporate at room temperature or 60°C; be careful not to let the gDNA overdry.
Resuspend gDNA in 30 uL dH2O plus 0.1 mg/mL RNase A per well, cover the plate with PCR plate film, and place on shaker at moderate speed (to avoid shearing of DNA) for about 2 hours. Leave at room temperature overnight and store at –20°C.
Screen hPSC clones by PCR amplification of the targeted genomic region: use primers that amplify a short product (150--200 bp) around the genomic target site; set up a PCR reaction for each hPSC clone using 96-well PCR plates and a multichannel pipette to pipette PCR master mix and gDNA; use 2.5 uL gDNA per 25 uL PCR reaction (consider increasing amount of template DNA if no PCR products are obtained); include a negative control PCR reaction (H
2O only); run PCR products on a high percentage (2.5%) agarose gel (load gel using a multichannel pipette) for 2--3 hours to separate wild type from indel-containing products (). If a restriction site was inserted into the genomic target site to facilitate screening (see
Section 3.1.7), digest 3 uL of the PCR reaction for each hPSC clone in a 20 uL restriction digest; PCR purification prior to restriction digestion is not necessary if the volume of the PCR reaction digested is kept relatively low; run the restriction digest on a high percentage (2.5%) agarose gel to separate undigested and digested PCR products.
Confirm suspected mutant clones by direct sequencing of the PCR products and sequencing of TOPO-cloned PCR products.
Clone recovery
Prepare Geltrex-coated 24-well plate (1 well each for mutant and control clones of interest).
Add 500--750 uL mTeSR1 per well plus 10 uM ROCK inhibitor (taking into account the final volume per well).
Remove frozen 96-well plate from –80°C freezer, clean outside of plate with 70% ethanol, remove Parafilm, and thaw quickly by placing the plate in a 37°C tissue-culture incubator (wells on the borders of the plate will thaw more quickly).
When wells of interest have only a small piece of ice remaining, move plate into a tissue-culture hood and add 100 uL room temperature mTeSR1 directly to each well.
Quickly transfer the thawed cells using a P200 pipette (using a new filter tip for each well) into the wells of the 24-well plate.
Replace the media with fresh mTeSR1 (without ROCK inhibitor) every 24 hours.
Once large colonies with hPSC morphology appear, expand to one well of a 6-well plate, and then expand to a 10-cm plate (some colonies will not thaw successfully and will be lost due to poor viability or differentiation, so it is recommended that several suspected mutant and control clones be thawed).
Materials
15-cm, 10-cm, 6-well, 24-well, and 96-well tissue culture plates (any source)
mTeSR1 media (STEMCELL Technologies)
Penicillin/streptomycin (Gibco or other source)
Geltrex matrix (Life Technologies)
Accutase (STEMCELL Technologies)
PBS, calcium- and magnesium-free (Gibco or other source)
bFGF (Aldevron)
Media filter bottles (VWR or other source)
Gentamicin reagent solution (Gibco or other source)
ROCK inhibitor, Y-27632 (Santa Cruz Biotechnologies, sc-281642A)
Electroporator (Bio-Rad or other source)
Electroporation cuvettes, 0.4 cm (Bio-Rad or other source)
FACS tube (BD Biosciences or other source)
mFreSR (STEMCELL Technologies)
Proteinase K (Roche or other source)
RNase A (Invitrogen or other source)
PCR plate film (VWR or other source)
TOPO TA Cloning Kit (Invitrogen)
Troubleshooting
Low percentage of GFP- and/or RFP-positive cells after electroporation
Unknown factors lead to variable percentages of GFP- and/or RFP-positive cells from one electroporation attempt to another, and the percentage of GFP- and/or RFP-positive cells may be low (<5% GFP-positive and nearly 0% RFP-positive, see Section 5.3.2). However, we have not noticed a correlation between the percentage of fluorescent cells and the efficiency of genome editing using this protocol. Therefore, the observed fluorescence percentages are not important if a suitable number of cells can be collected for clonal plating. Although the fluorescent cell percentage may not be important, note the following: 15,000 to 50,000 cells are needed for post-FACS clonal plating per 10-cm plate (see Section 5.1.3); it is important that the cells being collected are truly fluorescent (see gating strategy illustrated in ); and FACS should be completed relatively quickly (less than 1.5 hours of total FACS time, see Section 5.3.3) to avoid reduced cell viability upon clonal plating.
The percentage of GFP- and/or RFP-positive cells may be influenced by the rate of cell division after electroporation, which may be greater for some hPSC lines; cell division after electroporation appears to result in a lower GFP/RFP-positive percentage during FACS, likely due to dilution of plasmid DNA and GFP/RFP as the cells divide.
Very low percentage of RFP-positive cells after electroporation
For unclear reasons, the percentage of RFP-positive and GFP/RFP-double-positive cells during FACS is typically an order of magnitude less than the percentage of GFP-positive cells (see ). This discrepancy does not affect the efficiency of genome editing. GFP-positive, RFP-positive, and GFP/RFP-double-positive cells can be collected into a single collection tube (see Section 5.1.3).
Few or no surviving colonies on post-FACS 10-cm plate
As described in Section 5.1.3, different hPSC lines exhibit different survival rates during clonal plating to 10-cm plates after FACS, necessitating some optimization of plating density for a given cell line (generally ranging from 15,000 to 50,000 cells per 10-cm plate). Therefore, if no colonies survive clonal plating, consider plating at a higher density.
Conditioned media (see Section 5.1.1) increases the survival rate during clonal plating after FACS and should be used after FACS until small colonies appear (typically 2 to 4 days after FACS), at which point normal mTeSR1 media is used.
The post-FACS survival rate may decrease as the overall FACS time increases. It is best to complete the entire FACS-enrichment process (see Section 5.1.3)—beginning with collecting the cells and ending with plating to 10-cm plates—within 1.5 hours. If the FACS is proceeding slowing (due to a low percentage of fluorescent cells), consider plating the appropriate number of cells to a 10-cm plate once that amount of cells has been collected, while the FACS is still proceeding.
Bacterial contamination occurs on post-FACS 10-cm plates or 96-well plates
If bacterial contamination is observed on any of the post-FACS 10-cm plates or 96-well plates (likely due to contamination from the flow cytometer or the multichannel aspirator respectively), throw away the contaminated plates and add 50 ug/mL gentamicin to the media of any remaining plates.
Cell density in 96-well plates varies from well to well
After picking hPSC colonies to 96-well plates, the cell density may be appreciably different from well to well within an individual plate. If this is the case, some wells may be ready to be passaged (i.e., reach 80% confluence) before other wells. To avoid this problem, when picking colonies, choose colonies that are similar in size. When considering when to passage a 96-well plate where the cell density varies between wells, wait to passage the plate until the majority of the wells have reached a suitable level of confluence.
Screening PCR reactions fail for a significant number of hPSC clones
The PCR reaction should be optimized with control genomic DNA before setting up large-scale screening PCR reactions to ensure that PCR products will be obtained for most of the hPSC clones during the screening PCR. Failure of a significant number of PCR reactions during the large-scale screening PCR may occur if the PCR reaction is not well-optimized; if this is the case, you may observe small bright primer dimer bands on the gel. Setting up the screening PCR reactions on ice as quickly as possible will increase the chances of success.
Poor-quality or low-concentration template genomic DNA from most or all of the hPSC clones may cause the screening PCR reactions to fail. Ensure that the DNA is fully dissolved before setting up the screening PCR reactions; consider increasing the volume of template genomic DNA used.
Lastly, be careful not to lose the precipitated genomic DNA pellets from the wells of the 96-well plate when preparing genomic DNA (Section 5.1.5). Precipitating the genomic DNA for several hours at –20°C and spinning the plates down after precipitating the DNA helps diminish this outcome.
Conclusions
The protocols described here facilitate efficient genome editing in hPSCs using TALENs or Cas9/CRISPRs. Notably, our approach employs FACS following electroporation to isolate hPSCs that express TALENs or Cas9/CRISPRs (the mediators of genome editing), overcoming a significant obstacle to facile gene targeting in hPSCs: inefficient delivery of DNA. This overall approach is straightforward, takes only a few weeks in total, does not require antibiotic selection, and has a wide array of potential applications. Efficient genome editing greatly expands what is possible within the field of hPSC research.
Acknowledgements
This work was supported in part by the Sternlicht Director's Fund Award for Graduate Students from the Harvard Stem Cell Institute (D.T.P.); grants R00-HL098364 (K.M.), U01-HL107440 (C.A.C.), and R01-DK097768 (K.M., C.A.C.) from the United States National Institutes of Health (NIH); the Broad Institute's Lawrence H. Summers Fellowship and the Carlos Slim Foundation (K.M.); the Harvard Stem Cell Institute (K.M., C.A.C.), and Harvard University (K.M., C.A.C.). The authors report no relevant conflicts of interest. We thank J. Keith Joung, Feng Zhang, George Church, Prashant Mali, Qiurong Ding, Nicolas Kuperwasser, Jennifer Shay, and the staff of the HSCRB-HSCI Flow Cytometry Core for assistance and suggestions.
References
Cowan C.A, Klimanskaya I, McMahon J, Atienza J, Witmyer J, Zucker J.P, Wang S, Morton C.C, McMahon A.P, Powers D, Melton D.A. Derivation of embryonic stem-cell lines from human blastocysts.
N Engl J Med. 2004;350(13):1353–1356. [
PubMed: 14999088] [
CrossRef]
Ding Q, Lee Y.K, Schaefer E.A, Peters D.T, Veres A, Kim K, Kuperwasser N, Motola D.L, Meissner T.B, Hendriks W.T, Trevisan M, Gupta R.M, Moisan A, Banks E, Friesen M, Schinzel R.T, Xia F, Tang A, Xia Y, Figueroa E, Wann A, Ahfeldt T, Daheron L, Zhang F, Rubin L.L, Peng L.F, Chung R.T, Musunuru K, Cowan C.A. A TALEN genome-editing system for generating human stem cell-based disease models.
Cell Stem Cell. 2013a;12(2):238–251. [
PMC free article: PMC3570604] [
PubMed: 23246482] [
CrossRef]
Ding Q, Regan S.N, Xia Y, Oostrom L.A, Cowan C.A, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs.
Cell Stem Cell, http://dx.doi.org/ 10.1016/j.stem.2013.03.006. 2013b [
PMC free article: PMC3925309] [
PubMed: 23561441] [
CrossRef]
Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver R.C, Katibah G.E, Amora R, Boydston E.A, Zeitler B, Meng X, Miller J.C, Zhang L, Rebar E.J, Gregory P.D, Urnov F.D, Jaenisch R. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases.
Nat Biotechnol. 2009;27(9):851–857. [
PMC free article: PMC4142824] [
PubMed: 19680244] [
CrossRef]
Hockemeyer D, Wang H, Kiani S, Lai C.S, Gao Q, Cassady J.P, Cost G.J, Zhang L, Santiago Y, Miller J.C, Zeitler B, Cherone J.M, Meng X, Hinkley S.J, Rebar E.J, Gregory P.D, Urnov F.D, Jaenisch R. Genetic engineering of human pluripotent cells using TALE nucleases.
Nat Biotechnol. 2011;29(8):731–734. [
PMC free article: PMC3152587] [
PubMed: 21738127] [
CrossRef]
Last revised March 28, 2013. Published April 29, 2013. This chapter should be cited as: Peters, D.T., Cowan, C.A. and Musunuru, K., Genome editing in human pluripotent stem cells (April 29, 2013), StemBook, ed. The Stem Cell Research Community, StemBook, doi/10.3824/stembook.1.94.1.