Summary
Clinical characteristics.
Campomelic dysplasia (CD) is a skeletal dysplasia characterized by distinctive facies, Pierre Robin sequence with cleft palate, shortening and bowing of long bones, and clubfeet. Other findings include laryngotracheomalacia with respiratory compromise and ambiguous genitalia or normal female external genitalia in most individuals with a 46,XY karyotype. Many affected infants die in the neonatal period; additional findings identified in long-term survivors include short stature, cervical spine instability with cord compression, progressive scoliosis, and hearing impairment.
Management.
Treatment of manifestations: Care of children with cleft palate by a craniofacial team using routine measures; in persons with a 46,XY karyotype and undermasculinization of the genitalia, the gonads should be removed because of the increased risk for gonadoblastoma; care of hip subluxation and clubfeet using standard protocols; hearing aids for those with hearing impairment; surgery as needed for cervical vertebral instability and progressive cervicothoracic kyphoscoliosis that compromises lung function.
Prevention of secondary complications: If a cervical spine abnormality is identified, special care should be exercised for any surgical procedure.
Surveillance: Annual monitoring of spinal curvature.
Genetic counseling.
CD is inherited in an autosomal dominant manner. To date, most probands have CD as the result of a de novo pathogenic variant in SOX9; thus, parents of probands are not typically affected. However, a few adults have been diagnosed with CD following the birth of an affected child. Recurrence in sibs has occurred and somatic and germline mosaicism have been reported. Prenatal testing for a pregnancy at increased risk is possible if the pathogenic variant in the family is known.
Diagnosis
No consensus clinical diagnostic criteria for campomelic dysplasia (CD) have been published. The diagnosis of CD (derived from the Greek for "bent limb") can usually be clearly established based on clinical and radiographic findings. Although no single clinical feature is obligatory, the radiographic features are consistent and are the most reliable diagnostic clues.
Suggestive Findings
CD should be suspected in individuals with the following clinical and radiographic features.
Clinical features
Relatively large head
Pierre Robin sequence with cleft palate
Midface hypoplasia
Laryngotracheomalacia
Respiratory distress
Eleven pairs of ribs
Ambiguous genitalia or normal female external genitalia in an individual with a 46,XY
karyotypeDislocatable hips
Short bowed limbs (lower limbs more frequently than upper limbs)
Pretibial skin dimples (Bowing of the lower leg is often associated with a skin dimple over the apex of curve.)
Clubfeet
Note: Bowing of the limbs, the feature that gave the disorder its name, is neither specific nor an obligatory finding. When the limbs are not bowed, the term "acampomelic campomelic dysplasia" is used. Bowing of the limbs is present in many other skeletal dysplasias (e.g., osteogenesis imperfecta).
Radiographic findings (, , )
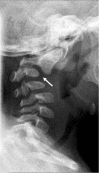
Cervical spine changes (i.e., abnormal AP curvature and anterior dislocation of C2 on C3) (arrow) in a boy age 11 months with classic campomelic dysplasia
Molecularly confirmed "acampomelic" campomelic dysplasia A. Tracheostomy tube is in place and the scapulae are markedly hypoplastic (arrows).
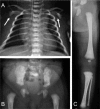
Classic radiographic features of campomelic dysplasia in a 24-week fetus. Note cervical spine abnormalities, hypoplastic thoracic vertebral pedicles, scapular hypoplasia, narrow iliac wings, bowing of the femora and the tibiae, and clubfeet.
Cervical spine anomalies (variable, often kyphosis) ()
Scapular hypoplasia (, )
Hypoplastic thoracic vertebral pedicles ()
Eleven pairs of ribs
Scoliosis or kyphoscoliosis
Vertically oriented narrow iliac wings ()
Bowed femora and/or tibiae (occasionally upper limb) ()
Establishing the Diagnosis
The clinical diagnosis of CD can be established in a proband with the clinical and radiographic findings described in Suggestive Findings. Identification of a heterozygous pathogenic (or likely pathogenic) variant in SOX9 by molecular genetic testing (see Table 1) can establish the diagnosis if clinical and radiographic findings are inconclusive.
The molecular diagnosis of CD is established in a proband with suggestive findings who has one of the following on molecular genetic testing (see Table 1):
A
heterozygous interstitial
deletion or reciprocal
translocation of 17q24.3-q25.1 involving
SOX9 or its regulatory region (5% of affected individuals) [G Scherer, unpublished data]
Note: In rare instances, the
translocation may be
familial; thus, parental karyotypes should be analyzed when an abnormality is found in the
proband.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a heterozygous SOX9 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel), chromosomal microarray, karyotype, and comprehensive
genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with skeletal dysplasia and/or ambiguous genitalia are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SOX9 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Chromosomal microarray analysis (CMA) uses oligonucleotide or SNP arrays to detect genome-wide large deletions/duplications (including SOX9) that cannot be detected by sequence analysis.
Karyotype. If SOX9 testing is not diagnostic, a karyotype may be considered to evaluate for a reciprocal translocation that involves 17q24.3-q25.1 (SOX9 locus) but does not result in SOX9 copy number changes. (See Management for recommendations regarding karyotype in phenotypic females with campomelic dysplasia.)
A skeletal dysplasia multigene panel that includes SOX9 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. The composition of the gene panel is likely to vary depending on presenting feature(s) and age at presentation (e.g., bowed limbs in a fetus, Pierre Robin sequence in a newborn). Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by skeletal dysplasia, comprehensive
genomic testing, which does not require the clinician to determine which gene is likely involved, is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic – and particularly when evidence supports autosomal dominant inheritance – exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Campomelic Dysplasia
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
SOX9
| Sequence analysis (incl 3' & 5'UTR) 3 | 90%-95% 4 |
| Gene-targeted deletion/duplication analysis 5, 6 | ~2% 7 |
| CMA 8 | ~1% 9 |
| Karyotype | ~1% 10 |
- 1.
- 2.
- 3.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications. Gene-targeted deletion/duplication testing will detect deletions ranging from a single exon to the whole gene; however, breakpoints of large deletions and/or deletion of adjacent genes (e.g., the family described by Castori et al [2016]) may not be detected by these methods.
- 6.
- 7.
- 8.
Chromosomal microarray analysis (CMA) uses oligonucleotide or SNP arrays to detect genome-wide large deletions/duplications (including SOX9) that cannot be detected by sequence analysis. The ability to determine the size of the deletion/duplication depends on the type of microarray used and the density of probes in the 17q24.3 region. CMA designs in current clinical use target the 17q24.3 region.
- 9.
- 10.
Clinical Characteristics
Clinical Description
To date, approximately 100 individuals (fetuses included) with a pathogenic variant in SOX9 have been identified; the data are scattered across many case reports and a few small series [Cameron et al 1996, Pfeifer et al 1999, Mansour et al 2002, Pop et al 2004, Hill-Harfe et al 2005, Smyk et al 2007, Lecointre et al 2009, Gentilin et al 2010, Corbani et al 2011, Fonseca et al 2013, Mattos et al 2015, Castori et al 2016, Csukasi et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Campomelic dysplasia (CD) is sometimes identified on prenatal ultrasound examination but may escape detection until after birth if the limbs are not bowed.
Many newborns with CD die shortly after birth secondary to respiratory insufficiency. In comparison with other lethal skeletal dysplasias, the cause of death in CD is not related to thoracic cage hypoplasia but rather to airway instability (tracheobronchomalacia) or cervical spine instability. Nonetheless, a number of infants with CD have survived the neonatal period [Mansour et al 2002].
The facies in CD resembles the type 2 collagen disorders (e.g., Stickler syndrome), with Pierre Robin sequence (with cleft palate) and flat midface. In the newborn period, the midface is hypoplastic and the eyes are prominent. Relatively large head size (in comparison to total body length) is common. The limbs are short with body length often below the third percentile. Bowing of the limbs is often present but not obligatory.
Approximately 75% of individuals with CD who have a 46,XY karyotype have either ambiguous external genitalia or normal female external genitalia. The internal genitalia are variable, often with a mixture of müllerian and wolffian duct structures.
Given the relatively small number of survivors described in the literature, it is difficult to generalize about the natural history. The following have been observed:
Intellect is normal.
Height is variably affected. Some newborns have significant short stature whereas others are within the normal range.
When present, scoliosis is usually progressive, contributes to the short stature, and may result in neurologic signs and symptoms.
Vertebral hypoplasia or malformation, particularly of the cervical spine, may lead to neurologic signs of cord compression unless surgically stabilized and may be the cause of death among those who initially survive the newborn period.
Hearing impairment/loss in some can be significant enough to require hearing aids.
A variety of
congenital heart defects have been reported in a minority of cases.
Histologic pancreatic abnormalities have been described in three newborns who died at term from CD; however, pancreatic dysfunction has not been seen in survivors with CD [
Piper et al 2002].
Ischiopubic-patella syndrome (IPP). The phenotypic description of IPP is limited to findings in the pelvis and legs including hypoplastic patellae, hypoplastic lesser trochanters, and defective ischiopubic ossification. In several persons with this diagnosis, pathogenic variants of SOX9 or cytogenetic alterations in the vicinity of SOX9 have been reported [Mansour et al 2002]. It is now recognized that individuals with IPP have a mild form of campomelic dysplasia with survival to adulthood.
Genotype-Phenotype Correlations
Clear-cut genotype-phenotype correlations are not readily apparent in CD [Meyer et al 1997]. However, correlations of some degree are observed in those with the following two findings.
Chromosomal rearrangements. In long-term survivors with CD and those with acampomelic campomelic dysplasia, de novo translocations or inversions with breakpoints upstream of SOX9 are more likely to be seen than pathogenic variants in the SOX9 coding region [Pfeifer et al 1999, Leipoldt et al 2007, Gordon et al 2009, Jakubiczka et al 2010, Fukami et al 2012]. In general, the farther the breakpoint is from SOX9, the milder the phenotype, including the effect on male external genitalia [Leipoldt et al 2007] and skeletal findings.
Acampomelic campomelic dysplasia (ACD). Mild campomelia and ACD are overrepresented in those with translocations or inversions, accounting for nine of 15 cases with well-defined breakpoints [Leipoldt et al 2007]. In contrast, only approximately 10% of individuals with pathogenic variants in the SOX9 coding region have ACD. Notably, these are mostly missense variants in the DNA-binding domain [Staffler et al 2010, Corbani et al 2011]. Furthermore, the single missense variant not located in this domain was located in the SOX9 dimerization domain in two unrelated individuals with ACD [Bernard et al 2003, Sock et al 2003]. In addition, the few individuals with SOX9 upstream deletions all had ACD [Pop et al 2004, Lecointre et al 2009, White et al 2011]. Thus, compared to individuals with CD, individuals with ACD have a significantly higher probability of having either a genomic rearrangement with breakpoint upstream of SOX9, a SOX9 upstream deletion, or a SOX9 missense variant.
Penetrance
Pathogenic variants in the SOX9 coding region are completely penetrant.
Breakpoints at long distance from SOX9 may not be completely penetrant.
Nomenclature
The name "campomelic dysplasia," first proposed by Maroteaux in 1971, is derived from the Greek for "bent limb." Other terms used in the past to refer to campomelic dysplasia include campomelic dwarfism, campomelic syndrome, and camptomelic dwarfism.
Although the name "campomelic dysplasia" is well established, it can lead to confusion, as not every child with CD has bowed limbs (ACD) and, conversely, most children with bowed limbs do not have CD but another of the frequent genetic disorders of bone, including osteogenesis imperfecta (OI), hypophosphatasia, cartilage-hair hypoplasia, and others (see Differential Diagnosis).
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], CD is referred to as SOX9-related campomelic dysplasia and included in the bent bones dysplasia group.
Prevalence
No reliable data exist regarding the prevalence of CD. The authors estimate it to be in the range of 1:40,000 to 1:80,000.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with campomelic dysplasia (CD), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended for those infants surviving the neonatal period.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Campomelic Dysplasia
View in own window
| System/Concern | Evaluation | Comment |
|---|
Respiratory distress due to
laryngotracheomalacia
or tracheobronchomalacia
| Clinical eval | |
|
Cervical spine instability
| Lateral radiograph of cervical spine | |
|
Cleft palate
| Eval by craniofacial team incl feeding eval | |
|
Risk of gonadoblastoma in 46,XY phenotypic females
| Karyotype analysis | In phenotypic females to identify those w/46,XY karyotype |
|
Clubfeet
| Referral to orthopedist | |
|
Hearing impairment
| Hearing screening | |
|
Genetic counseling
| By genetics professionals 1 | To inform affected persons & their families re nature, MOI, & implications of CD to facilitate medical & personal decision making |
CD = campomelic dysplasia
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with Campomelic Dysplasia
View in own window
| Manifestation/Concern | Treatment | Considerations/Other |
|---|
|
Cleft palate
| Care by craniofacial team & surgical closure | |
46,XY karyotype &
female genitalia
| Gonadectomy because of ↑ risk of gonadoblastoma | No data available re appropriate age for this procedure |
Hip dislocation/
luxation
| Treatment per orthopedist | |
|
Clubfeet
| Surgical correction per orthopedist | |
|
Hearing impairment
| Treatment per audiologist incl hearing aids | |
Progressive
cervicothoracic
kyphoscoliosis
| Surgical treatment per orthopedist/neurosurgeon | Surgery often required in childhood for those w/compromised lung function [Thomas et al 1997]; bracing usually not helpful |
Cervical spine
instability
| Surgical treatment per orthopedist/neurosurgeon | |
Prevention of Secondary Complications
Risk associated with use of anesthesia prior to imaging or surgery. If a cervical spine abnormality is identified, special care should be exercised for any surgical procedure.
Surveillance
Table 5.
Recommended Surveillance for Individuals with Campomelic Dysplasia
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Kyphoscoliosis
| Clinical & radiographic assessment for spinal curvature | Annually in long-term survivors |
Agents/Circumstances to Avoid
There are no known circumstances to avoid. However, in long-term survivors with cervical spine malformations, it seems reasonable to limit activities that cause extreme flexion or extension (e.g., somersaults).
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Risk to Family Members
Parents of a proband
To date, most probands with campomelic dysplasia (CD) have the disorder as the result of a
de novo
SOX9 pathogenic variant; thus, parents of a
proband are not typically affected.
Genetic testing capable of detecting the
SOX9 pathogenic variant or
chromosome rearrangement identified in the
proband is recommended for the parents of a proband to confirm the genetic status of the parents and to allow reliable
recurrence risk assessment. (Note: Familial translocations have been reported but are rare.)
Sibs of a proband. The risk to the sibs of a proband depends on the genetic status of the proband's parents:
If neither parent has a
chromosome rearrangement, the risk to sibs is negligible.
If a parent has a balanced
chromosome rearrangement, the risk to sibs is increased and depends on the specific chromosome rearrangement and the possibility of other variables.
Offspring of a proband. Many individuals with CD do not survive infancy; some, however, have reproduced.
Each child of an individual with a non-mosaic
SOX9 pathogenic variant has a 50% chance of inheriting the pathogenic variant.
The risk to offspring of an individual with a
chromosome rearrangement involving
SOX9 depends on the
cytogenetic abnormality.
Other family members. The risk to other family members depends on the status of the proband's parents:
If a parent has a balanced
chromosome rearrangement, the parent's family members are at risk and can be offered chromosome analysis.
Prenatal Testing and Preimplantation Genetic Testing
A priori high-risk pregnancies
Similarly,
prenatal testing for a pregnancy at increased risk for a
familial chromosome rearrangement is possible by chromosome analysis of fetal cells obtained by amniocentesis or chorionic villus sampling.
A priori low-risk pregnancies. Routine prenatal ultrasound examination may identify skeletal findings (e.g., increased nuchal translucency, micrognathia, short bowed limbs, and hypoplastic scapulae) that raise the possibility of CD in a fetus not known to be at increased risk [Schramm et al 2009, Gentilin et al 2010]. Once a skeletal dysplasia is identified prenatally, it is often difficult to establish the diagnosis based on ultrasound findings alone. Consideration of molecular genetic testing for a SOX9 pathogenic variant in these situations is appropriate; multigene panel testing or exome sequencing may be more efficient in the setting of nonspecific ultrasound signs such as bowed limbs or midface hypoplasia.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Campomelic Dysplasia: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
Pathogenic variants within the SOX9 coding region lead to an altered SOX9 protein with impaired activity to function as a transcription factor. In contrast, chromosome rearrangements (translocations, inversions) with breakpoints as far as ~1 Mb upstream of SOX9 as well as SOX9 upstream deletions leave the SOX9 coding region intact but most likely lead to reduced expression of SOX9 by interrupting its extended cis-regulatory domain. In either case, SOX9 function as a developmental regulator is compromised.
SOX9 is a proven key regulator at various steps of chondrocyte differentiation, regulating expression of the collagen genes COL2A1 and COL11A2 as well as of CD-RAP and ACAN (also known as AGGRECAN) [Akiyama & Lefebvre 2011].
Regulation of COL2A1 by SOX9 may explain some of the phenotypic overlap of campomelic dysplasia (CD) with spondyloepiphyseal dysplasia congenita.
Studies in mice provide evidence that the murine ortholog of human SOX9 also plays a role during formation of the pancreas, heart, gut, and inner ear.
Thus, the wide spectrum of pathologic symptoms seen in CD, including the skeletal defects, XY sex reversal, pancreatic defects (size reduction of islets of Langerhans and reduced insulin secretion), heart defects, and sensorineural and conductive hearing impairment, can be attributed directly to impaired ability of the pleiotropic developmental regulator SOX9 to activate target genes during organogenesis.
Mechanism of disease causation. Pathogenic nonsense and most frameshift variants in SOX9 predict a prematurely truncated protein resulting in loss-of-function alleles. SOX9 pathogenic variants that result in a mutated protein retaining the HMG domain (a DNA-binding domain) may function as dominant-negative alleles.