Summary
Clinical characteristics.
Campomelic dysplasia (CD) is a skeletal dysplasia characterized by distinctive facies, Pierre Robin sequence with cleft palate, shortening and bowing of long bones, and clubfeet. Other findings include laryngotracheomalacia with respiratory compromise and ambiguous genitalia or normal female external genitalia in most individuals with a 46,XY karyotype. Many affected infants die in the neonatal period; additional findings identified in long-term survivors include short stature, cervical spine instability with cord compression, progressive scoliosis, and hearing impairment.
Diagnosis/testing.
The diagnosis of CD is usually based on clinical and radiographic findings. Identification of a heterozygous pathogenic variant in SOX9 by molecular genetic testing can confirm the diagnosis if clinical and radiographic features are inconclusive.
Management.
Treatment of manifestations: Care of children with cleft palate by a craniofacial team using routine measures; in persons with a 46,XY karyotype and undermasculinization of the genitalia, the gonads should be removed because of the increased risk for gonadoblastoma; care of hip subluxation and clubfeet using standard protocols; hearing aids for those with hearing impairment; surgery as needed for cervical vertebral instability and progressive cervicothoracic kyphoscoliosis that compromises lung function.
Prevention of secondary complications: If a cervical spine abnormality is identified, special care should be exercised for any surgical procedure.
Surveillance: Annual monitoring of spinal curvature.
Genetic counseling.
CD is inherited in an autosomal dominant manner. To date, most probands have CD as the result of a de novo pathogenic variant in SOX9; thus, parents of probands are not typically affected. However, a few adults have been diagnosed with CD following the birth of an affected child. Recurrence in sibs has occurred and somatic and germline mosaicism have been reported. Prenatal testing for a pregnancy at increased risk is possible if the pathogenic variant in the family is known.
GeneReview Scope
Table
Acampomelic campomelic dysplasia Campomelic dysplasia
Diagnosis
No consensus clinical diagnostic criteria for campomelic dysplasia (CD) have been published. The diagnosis of CD (derived from the Greek for "bent limb") can usually be clearly established based on clinical and radiographic findings. Although no single clinical feature is obligatory, the radiographic features are consistent and are the most reliable diagnostic clues.
Suggestive Findings
CD should be suspected in individuals with the following clinical and radiographic features.
Clinical features
- Relatively large head
- Pierre Robin sequence with cleft palate
- Midface hypoplasia
- Laryngotracheomalacia
- Respiratory distress
- Eleven pairs of ribs
- Ambiguous genitalia or normal female external genitalia in an individual with a 46,XY karyotype
- Dislocatable hips
- Short bowed limbs (lower limbs more frequently than upper limbs)
- Pretibial skin dimples (Bowing of the lower leg is often associated with a skin dimple over the apex of curve.)
- Clubfeet
Note: Bowing of the limbs, the feature that gave the disorder its name, is neither specific nor an obligatory finding. When the limbs are not bowed, the term "acampomelic campomelic dysplasia" is used. Bowing of the limbs is present in many other skeletal dysplasias (e.g., osteogenesis imperfecta).
Radiographic findings (Figure 1, Figure 2, Figure 3)
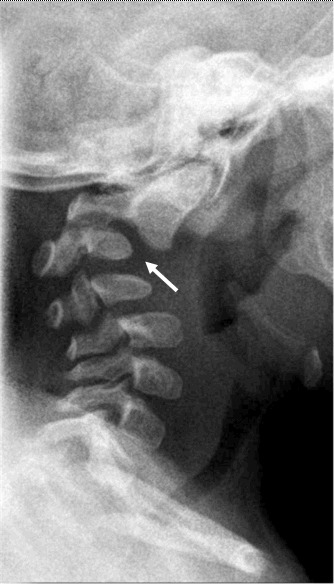
Figure 1.
Cervical spine changes (i.e., abnormal AP curvature and anterior dislocation of C2 on C3) (arrow) in a boy age 11 months with classic campomelic dysplasia
Figure 2.
Molecularly confirmed "acampomelic" campomelic dysplasia A. Tracheostomy tube is in place and the scapulae are markedly hypoplastic (arrows).

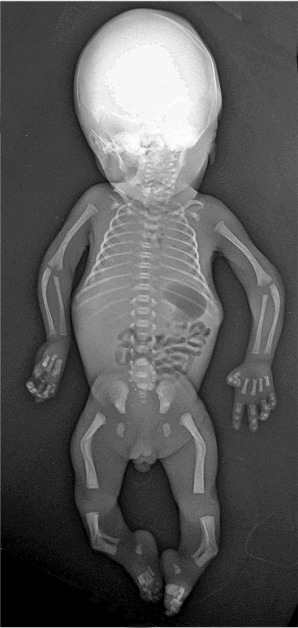
Figure 3.
Classic radiographic features of campomelic dysplasia in a 24-week fetus. Note cervical spine abnormalities, hypoplastic thoracic vertebral pedicles, scapular hypoplasia, narrow iliac wings, bowing of the femora and the tibiae, and clubfeet.
Establishing the Diagnosis
The clinical diagnosis of CD can be established in a proband with the clinical and radiographic findings described in Suggestive Findings. Identification of a heterozygous pathogenic (or likely pathogenic) variant in SOX9 by molecular genetic testing (see Table 1) can establish the diagnosis if clinical and radiographic findings are inconclusive.
The molecular diagnosis of CD is established in a proband with suggestive findings who has one of the following on molecular genetic testing (see Table 1):
- A heterozygous pathogenic (or likely pathogenic) variant involving SOX9 (~90% of affected individuals) [Pop et al 2004; G Scherer, unpublished data]
- A heterozygous interstitial deletion or reciprocal translocation of 17q24.3-q25.1 involving SOX9 or its regulatory region (5% of affected individuals) [G Scherer, unpublished data]Note: In rare instances, the translocation may be familial; thus, parental karyotypes should be analyzed when an abnormality is found in the proband.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a heterozygous SOX9 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel), chromosomal microarray, karyotype, and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with skeletal dysplasia and/or ambiguous genitalia are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SOX9 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Chromosomal microarray analysis (CMA) uses oligonucleotide or SNP arrays to detect genome-wide large deletions/duplications (including SOX9) that cannot be detected by sequence analysis.
Karyotype. If SOX9 testing is not diagnostic, a karyotype may be considered to evaluate for a reciprocal translocation that involves 17q24.3-q25.1 (SOX9 locus) but does not result in SOX9 copy number changes. (See Management for recommendations regarding karyotype in phenotypic females with campomelic dysplasia.)
A skeletal dysplasia multigene panel that includes SOX9 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. The composition of the gene panel is likely to vary depending on presenting feature(s) and age at presentation (e.g., bowed limbs in a fetus, Pierre Robin sequence in a newborn). Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by skeletal dysplasia, comprehensive genomic testing, which does not require the clinician to determine which gene is likely involved, is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic – and particularly when evidence supports autosomal dominant inheritance – exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Campomelic Dysplasia
Clinical Characteristics
Clinical Description
To date, approximately 100 individuals (fetuses included) with a pathogenic variant in SOX9 have been identified; the data are scattered across many case reports and a few small series [Cameron et al 1996, Pfeifer et al 1999, Mansour et al 2002, Pop et al 2004, Hill-Harfe et al 2005, Smyk et al 2007, Lecointre et al 2009, Gentilin et al 2010, Corbani et al 2011, Fonseca et al 2013, Mattos et al 2015, Castori et al 2016, Csukasi et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Campomelic dysplasia (CD) is sometimes identified on prenatal ultrasound examination but may escape detection until after birth if the limbs are not bowed.
Many newborns with CD die shortly after birth secondary to respiratory insufficiency. In comparison with other lethal skeletal dysplasias, the cause of death in CD is not related to thoracic cage hypoplasia but rather to airway instability (tracheobronchomalacia) or cervical spine instability. Nonetheless, a number of infants with CD have survived the neonatal period [Mansour et al 2002].
The facies in CD resembles the type 2 collagen disorders (e.g., Stickler syndrome), with Pierre Robin sequence (with cleft palate) and flat midface. In the newborn period, the midface is hypoplastic and the eyes are prominent. Relatively large head size (in comparison to total body length) is common. The limbs are short with body length often below the third percentile. Bowing of the limbs is often present but not obligatory.
Approximately 75% of individuals with CD who have a 46,XY karyotype have either ambiguous external genitalia or normal female external genitalia. The internal genitalia are variable, often with a mixture of müllerian and wolffian duct structures.
Given the relatively small number of survivors described in the literature, it is difficult to generalize about the natural history. The following have been observed:
- Intellect is normal.
- Height is variably affected. Some newborns have significant short stature whereas others are within the normal range.
- When present, scoliosis is usually progressive, contributes to the short stature, and may result in neurologic signs and symptoms.
- Vertebral hypoplasia or malformation, particularly of the cervical spine, may lead to neurologic signs of cord compression unless surgically stabilized and may be the cause of death among those who initially survive the newborn period.
- Hearing impairment/loss in some can be significant enough to require hearing aids.
- A variety of congenital heart defects have been reported in a minority of cases.
- Histologic pancreatic abnormalities have been described in three newborns who died at term from CD; however, pancreatic dysfunction has not been seen in survivors with CD [Piper et al 2002].
Ischiopubic-patella syndrome (IPP). The phenotypic description of IPP is limited to findings in the pelvis and legs including hypoplastic patellae, hypoplastic lesser trochanters, and defective ischiopubic ossification. In several persons with this diagnosis, pathogenic variants of SOX9 or cytogenetic alterations in the vicinity of SOX9 have been reported [Mansour et al 2002]. It is now recognized that individuals with IPP have a mild form of campomelic dysplasia with survival to adulthood.
Genotype-Phenotype Correlations
Clear-cut genotype-phenotype correlations are not readily apparent in CD [Meyer et al 1997]. However, correlations of some degree are observed in those with the following two findings.
Chromosomal rearrangements. In long-term survivors with CD and those with acampomelic campomelic dysplasia, de novo translocations or inversions with breakpoints upstream of SOX9 are more likely to be seen than pathogenic variants in the SOX9 coding region [Pfeifer et al 1999, Leipoldt et al 2007, Gordon et al 2009, Jakubiczka et al 2010, Fukami et al 2012]. In general, the farther the breakpoint is from SOX9, the milder the phenotype, including the effect on male external genitalia [Leipoldt et al 2007] and skeletal findings.
- In two individuals with very distal translocation breakpoints (at 899 kb and 932 kb), the skeletal findings were so mild that they were transmitted through several generations [Hill-Harfe et al 2005, Velagaleti et al 2005].
- Misregulation of SOX9 has been implicated in individuals with isolated Pierre Robin sequence and translocation breakpoints 1.13 Mb upstream of SOX9 [Jakobsen et al 2007, Benko et al 2009, Gordon et al 2009, Fukami et al 2012].
Acampomelic campomelic dysplasia (ACD). Mild campomelia and ACD are overrepresented in those with translocations or inversions, accounting for nine of 15 cases with well-defined breakpoints [Leipoldt et al 2007]. In contrast, only approximately 10% of individuals with pathogenic variants in the SOX9 coding region have ACD. Notably, these are mostly missense variants in the DNA-binding domain [Staffler et al 2010, Corbani et al 2011]. Furthermore, the single missense variant not located in this domain was located in the SOX9 dimerization domain in two unrelated individuals with ACD [Bernard et al 2003, Sock et al 2003]. In addition, the few individuals with SOX9 upstream deletions all had ACD [Pop et al 2004, Lecointre et al 2009, White et al 2011]. Thus, compared to individuals with CD, individuals with ACD have a significantly higher probability of having either a genomic rearrangement with breakpoint upstream of SOX9, a SOX9 upstream deletion, or a SOX9 missense variant.
Penetrance
Pathogenic variants in the SOX9 coding region are completely penetrant.
Breakpoints at long distance from SOX9 may not be completely penetrant.
Nomenclature
The name "campomelic dysplasia," first proposed by Maroteaux in 1971, is derived from the Greek for "bent limb." Other terms used in the past to refer to campomelic dysplasia include campomelic dwarfism, campomelic syndrome, and camptomelic dwarfism.
Although the name "campomelic dysplasia" is well established, it can lead to confusion, as not every child with CD has bowed limbs (ACD) and, conversely, most children with bowed limbs do not have CD but another of the frequent genetic disorders of bone, including osteogenesis imperfecta (OI), hypophosphatasia, cartilage-hair hypoplasia, and others (see Differential Diagnosis).
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], CD is referred to as SOX9-related campomelic dysplasia and included in the bent bones dysplasia group.
Prevalence
No reliable data exist regarding the prevalence of CD. The authors estimate it to be in the range of 1:40,000 to 1:80,000.
Genetically Related (Allelic) Disorders
Isolated Pierre Robin sequence. Although it is likely to be rare, chromosomal translocations in the vicinity of SOX9 may cause isolated Pierre Robin sequence without other obvious findings of CD [Jakobsen et al 2007, Benko et al 2009, Gordon et al 2009].
Isolated sex reversal. There is a report of three adult sibs who were phenotypic females but had male karyotypes. There are no radiographs provided with the report, but the sex reversal is described as isolated. All three sibs had inherited a small 17q24 deletion (not seen in their two phenotypic 46,XX sisters) from their father [Bhagavath et al 2014].
Differential Diagnosis
Differential Diagnosis in the Prenatal Period
Table 2.
Disorders with Prenatal Limb Bowing in the Differential Diagnosis of Campomelic Dysplasia
Differential Diagnosis After Birth
After birth, the differential diagnosis is mainly spondyloepiphyseal dysplasia congenita (SEDC; see Type 2 Collagen Disorders Overview) because of the facial features, cleft palate, and short limbs. The milder type 2 collagenopathy Stickler syndrome may also be considered in the differential diagnosis as the facial features are very similar. Both disorders are caused by pathogenic variants in COL2A1 and inherited in an autosomal dominant manner. Radiographs differentiate these conditions.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with campomelic dysplasia (CD), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended for those infants surviving the neonatal period.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Campomelic Dysplasia
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with Campomelic Dysplasia
Prevention of Secondary Complications
Risk associated with use of anesthesia prior to imaging or surgery. If a cervical spine abnormality is identified, special care should be exercised for any surgical procedure.
Surveillance
Table 5.
Recommended Surveillance for Individuals with Campomelic Dysplasia
Agents/Circumstances to Avoid
There are no known circumstances to avoid. However, in long-term survivors with cervical spine malformations, it seems reasonable to limit activities that cause extreme flexion or extension (e.g., somersaults).
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Campomelic dysplasia (CD) is an autosomal dominant disorder typically caused by a de novo SOX9 pathogenic variant. Rarely, CD is the result of a de novo or inherited chromosome rearrangement (e.g., deletion, translocation, or inversion) upstream to or involving SOX9.
Risk to Family Members
Parents of a proband
- To date, most probands with campomelic dysplasia (CD) have the disorder as the result of a de novo SOX9 pathogenic variant; thus, parents of a proband are not typically affected.
- A few adults have been diagnosed with CD following the birth of an affected child [Mansour et al 2002, Savarirayan et al 2003, Lecointre et al 2009].
- Genetic testing capable of detecting the SOX9 pathogenic variant or chromosome rearrangement identified in the proband is recommended for the parents of a proband to confirm the genetic status of the parents and to allow reliable recurrence risk assessment. (Note: Familial translocations have been reported but are rare.)
- If the SOX9 pathogenic variant or chromosome rearrangement found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo alteration in the proband or somatic and/or germline mosaicism in a parent.
- Somatic and germline mosaicism in mildly affected parents of affected sibs has been reported in two families [Higeta et al 2018, Smyk et al 2007].
- Somatic and/or germline mosaicism for a SOX9 pathogenic variant has also been reported in unaffected parents [Wagner et al 1994, Cameron et al 1996, Gentilin et al 2010].
Sibs of a proband. The risk to the sibs of a proband depends on the genetic status of the proband's parents:
- If a parent of the proband is heterozygous for a SOX9 pathogenic variant identified in the proband, the risk to the sibs is 50%.
- If the proband has a known SOX9 pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 2%-5% because of the possibility of parental mosaicism [Wagner et al 1994, Cameron et al 1996, Smyk et al 2007, Gentilin et al 2010, Higeta et al 2018].
- If a proband has a chromosome rearrangement, the recurrence risk to sibs depends on the chromosome findings in the parents:
- If neither parent has a chromosome rearrangement, the risk to sibs is negligible.
- If a parent has a balanced chromosome rearrangement, the risk to sibs is increased and depends on the specific chromosome rearrangement and the possibility of other variables.
Offspring of a proband. Many individuals with CD do not survive infancy; some, however, have reproduced.
- Each child of an individual with a non-mosaic SOX9 pathogenic variant has a 50% chance of inheriting the pathogenic variant.
- The risk to offspring of an individual with a chromosome rearrangement involving SOX9 depends on the cytogenetic abnormality.
Other family members. The risk to other family members depends on the status of the proband's parents:
- Because CD typically occurs as a de novo SOX9 pathogenic variant, risk to other family members is presumed to be low.
- If a parent has a balanced chromosome rearrangement, the parent's family members are at risk and can be offered chromosome analysis.
Related Genetic Counseling Issues
Family planning. The optimal time for determination of genetic risk and discussion of the availability of prenatal testing is before pregnancy.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
A priori high-risk pregnancies
- Once a SOX9 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
- Similarly, prenatal testing for a pregnancy at increased risk for a familial chromosome rearrangement is possible by chromosome analysis of fetal cells obtained by amniocentesis or chorionic villus sampling.
A priori low-risk pregnancies. Routine prenatal ultrasound examination may identify skeletal findings (e.g., increased nuchal translucency, micrognathia, short bowed limbs, and hypoplastic scapulae) that raise the possibility of CD in a fetus not known to be at increased risk [Schramm et al 2009, Gentilin et al 2010]. Once a skeletal dysplasia is identified prenatally, it is often difficult to establish the diagnosis based on ultrasound findings alone. Consideration of molecular genetic testing for a SOX9 pathogenic variant in these situations is appropriate; multigene panel testing or exome sequencing may be more efficient in the setting of nonspecific ultrasound signs such as bowed limbs or midface hypoplasia.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MAGIC FoundationPhone: 630-836-8200Email: contactus@magicfoundation.org
- Human Growth Foundation
- Little People of AmericaPhone: 888-LPA-2001; 714-368-3689Fax: 707-721-1896Email: info@lpaonline.org
- Skeletal Dysplasia Group FreiburgUniversity Hospital Freiberg, Centre for Pediatrics and Adolescent MedicineMathildenstrasse 1Freiburg 79106GermanyPhone: 001-497-6127 0-4363Email: violetta.volz@uniklinik-freiburg.de
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Campomelic Dysplasia: Genes and Databases
Table B.
OMIM Entries for Campomelic Dysplasia (View All in OMIM)
Molecular Pathogenesis
Pathogenic variants within the SOX9 coding region lead to an altered SOX9 protein with impaired activity to function as a transcription factor. In contrast, chromosome rearrangements (translocations, inversions) with breakpoints as far as ~1 Mb upstream of SOX9 as well as SOX9 upstream deletions leave the SOX9 coding region intact but most likely lead to reduced expression of SOX9 by interrupting its extended cis-regulatory domain. In either case, SOX9 function as a developmental regulator is compromised.
SOX9 is a proven key regulator at various steps of chondrocyte differentiation, regulating expression of the collagen genes COL2A1 and COL11A2 as well as of CD-RAP and ACAN (also known as AGGRECAN) [Akiyama & Lefebvre 2011].
- Regulation of COL2A1 by SOX9 may explain some of the phenotypic overlap of campomelic dysplasia (CD) with spondyloepiphyseal dysplasia congenita.
- SOX9 functions as a testis-determining gene downstream of SRY, inducing the formation of Sertoli cells and production of the anti-müllerian hormone AMH (also known as MIS) [Vidal et al 2001]. Of note, duplication or deletion of a common region ~0.5 Mb upstream of SOX9 causing isolated disorders of sexual development in the absence of any CD symptoms have been reported [Benko et al 2011, Cox et al 2011, Vetro et al 2011].
- Studies in mice provide evidence that the murine ortholog of human SOX9 also plays a role during formation of the pancreas, heart, gut, and inner ear.
Thus, the wide spectrum of pathologic symptoms seen in CD, including the skeletal defects, XY sex reversal, pancreatic defects (size reduction of islets of Langerhans and reduced insulin secretion), heart defects, and sensorineural and conductive hearing impairment, can be attributed directly to impaired ability of the pleiotropic developmental regulator SOX9 to activate target genes during organogenesis.
Mechanism of disease causation. Pathogenic nonsense and most frameshift variants in SOX9 predict a prematurely truncated protein resulting in loss-of-function alleles. SOX9 pathogenic variants that result in a mutated protein retaining the HMG domain (a DNA-binding domain) may function as dominant-negative alleles.
Chapter Notes
Revision History
- 6 April 2023 (sw) Revision: "SOX9-Related Campomelic Dysplasia" added as a synonym; Nosology of Genetic Skeletal Disorders: 2023 Revision [Unger et al 2023] added to Nomenclature
- 18 March 2021 (sw) Comprehensive update posted live
- 9 May 2013 (me) Comprehensive update posted live
- 31 July 2008 (cg) Review posted live
- 14 May 2008 (su) Original submission
References
Literature Cited
- Akiyama H, Lefebvre V. Unraveling the transcriptional regulatory machinery in chondrogenesis. J Bone Miner Metab. 2011;29:390–5. [PMC free article: PMC3354916] [PubMed: 21594584]
- Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J, Jamshidi N, Essafi A, Heaney S, Gordon CT, McBride D, Golzio C, Fisher M, Perry P, Abadie V, Ayuso C, Holder-Espinasse M, Kilpatrick N, Lees MM, Picard A, Temple IK, Thomas P, Vazquez MP, Vekemans M, Roest Crollius H, Hastie ND, Munnich A, Etchevers HC, Pelet A, Farlie PG, Fitzpatrick DR, Lyonnet S. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet. 2009;41:359–64. [PubMed: 19234473]
- Benko S, Gordon CT, Mallet D, Sreenivasan R, Thauvin-Robinet C, Brendehaug A, Thomas S, Bruland O, David M, Nicolino M, Labalme A, Sanlaville D, Callier P, Malan V, Huet F, Molven A, Dijoud F, Munnich A, Faivre L, Amiel J, Harley V, Houge G, Morel Y, Lyonnet S. Disruption of a long-distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet. 2011;48:825–30. [PubMed: 22051515]
- Bernard P, Tang P, Liu S, Dewing P, Harley VR, Vilain E. Dimerization of SOX9 is required for chondrogenesis, but not for sex determination. Hum Mol Genet. 2003;12:1755–65. [PubMed: 12837698]
- Bhagavath B, Layman LC, Ullmann R, Shen Y, Ha K, Rehman K, Looney S, McDonough PG, Kim HG, Carr BR. Familial 46,XY sex reversal without campomelic dysplasia caused by a deletion upstream of the SOX9 gene. Mol Cell Endocrinol. 2014;393:1–7. [PMC free article: PMC4332518] [PubMed: 24907458]
- Cameron FJ, Hageman RM, Cooke-Yarborough C, Kwok C, Goodwin LL, Sillence DO, Sinclair AH. A novel germ line mutation in SOX9 causes familial campomelic dysplasia and sex reversal. Hum Mol Genet. 1996;5:1625–30. [PubMed: 8894698]
- Castori M, Bottillo I, Morlino S, Barone C, Cascone P, Grammatico P, Laino L, et al. Variability in a three-generation family with Pierre Robin sequence, acampomelic campomelic dysplasia, and intellectual disability due to a novel ∼1 Mb deletion upstream of SOX9, and including KCNJ2 and KCNJ16. Birth Defects Res A Clin Mol Teratol. 2016;106:61–8. [PubMed: 26663529]
- Corbani S, Chouery E, Eid B, Jalkh N, Ghoch JA, Mégarbané A. Mild campomelic dysplasia: report on a case and review. Mol Syndromol. 2011;1:163–8. [PMC free article: PMC3042119] [PubMed: 21373255]
- Cox JJ, Willatt L, Homfray T, Woods CG. A SOX9 duplication and familial 46,XX developmental testicular disorder. N Engl J Med. 2011;364:91–3. [PubMed: 21208124]
- Csukasi F, Duran I, Zhang W, Martin JH, Barad M, Bamshad M, Weis MA, Eyre D, Krakow D, Cohn DH. Dominant-negative SOX9 mutations in campomelic dysplasia. Hum Mutat. 2019;40:2344–52. [PMC free article: PMC7608528] [PubMed: 31389106]
- Fonseca AC, Bonaldi A, Bertola DR, Kim CA, Otto PA, Vianna-Morgante AM. The clinical impact of chromosomal rearrangements with breakpoints upstream of the SOX9 gene: two novel de novo balanced translocations associated with acampomelic campomelic dysplasia. BMC Med Genet. 2013;14:50. [PMC free article: PMC3658899] [PubMed: 23648064]
- Fukami M, Tsuchiya T, Takada S, Kanbara A, Asahara H, Igarashi A, Kamiyama Y, Nishimura G, Ogata T. Complex genomic rearrangement in the SOX9 5' region in a patient with Pierre Robin sequence and hypoplastic left scapula. Am J Med Genet. 2012;158A:1529–34. [PubMed: 22529047]
- Gentilin B, Forzano F, Bedeschi MF, Rizzuti T, Faravelli F, Izzi C, Lituania M, Rodriguez-Perez C, Bondioni MP, Savoldi G, Grosso E, Botta G, Viora E, Baffico AM, Lalatta F. Phenotype of five cases of prenatally diagnosed campomelic dysplasia harboring novel mutations of the SOX9 gene. Ultrasound Obstet Gynecol. 2010;36:315–23. [PubMed: 20812307]
- Gordon CT, Tan TY, Benko S, Fitzpatrick D, Lyonnet S, Farlie PG. Long-range regulation at the SOX9 locus in development and disease. J Med Genet. 2009;46:649–56. [PubMed: 19473998]
- Higeta D, Yamaguchi R, Takagi T, Nishimura G, Sameshima K, Saito K, Minegishi T. Familial campomelic dysplasia due to maternal germinal mosaicism. Congenit Anom (Kyoto). 2018;58:194–7. [PubMed: 29542186]
- Hill-Harfe KL, Kaplan L, Stalker HJ, Zori RT, Pop R, Scherer G, Wallace MR. Fine mapping of chromosome 17 translocation breakpoints > or = 900 kb upstream of SOX9 in acampomelic campomelic dysplasia and a mild, familial skeletal dysplasia. Am J Hum Genet. 2005;76:663–71. [PMC free article: PMC1199303] [PubMed: 15717285]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jakobsen LP, Ullmann R, Christensen SB, Jensen KE, Molsted K, Henriksen KF, Hansen C, Knudsen MA, Larsen LA, Tommerup N, Tumer Z. Pierre Robin sequence may be caused by dysregulation of SOX9 and KCNJ2. J Med Genet. 2007;44:381–6. [PMC free article: PMC2740883] [PubMed: 17551083]
- Jakubiczka S, Schröder C, Ullmann R, Volleth M, Ledig S, Gilberg E, Kroisel P, Wieacker P. Translocation and deletion around SOX9 in a patient with acampomelic campomelic dysplasia and sex reversal. Sex Dev. 2010;4:143–9. [PubMed: 20453475]
- Kayhan G, Calis P, Karcaaltincaba D, Tug E. Prenatal diagnosis of campomelic dysplasia due to SOX9 deletion. J Obstet Gynaecol. 2019;39:1175–6. [PubMed: 31234679]
- Lecointre C, Pichon O, Hamel A, Heloury Y, Michel-Calemard L, Morel Y, David A, Le Caignec C. Familial acampomelic form of campomelic dysplasia caused by a 960 kb deletion upstream of SOX9. Am J Med Genet. 2009;149A:1183–9. [PubMed: 19449405]
- Leipoldt M, Erdel M, Bien-Willner GA, Smyk M, Theurl M, Yatsenko S, Lupski JR, Lane AH, Shanske AL, Stankiewicz P, Scherer G. Two novel translocation breakpoints upstream of SOX9 define borders of the proximal and distal breakpoint cluster region in campomelic dysplasia. Clin Genet. 2007;71:67–75. [PubMed: 17204049]
- Mansour S, Offiah AC, McDowall S, Sim P, Tolmie J, Hall C. The phenotype of survivors of campomelic dysplasia. J Med Genet. 2002;39:597–602. [PMC free article: PMC1735206] [PubMed: 12161603]
- Mattos EP, Sanseverino MT, Magalhães JA, Leite JC, Félix TM, Todeschini LA, Cavalcanti DP, Schüler-Faccini L. Clinical and molecular characterization of a Brazilian cohort of campomelic dysplasia patients, and identification of seven new SOX9 mutations. Genet Mol Biol. 2015;38:14–20. [PMC free article: PMC4415563] [PubMed: 25983619]
- Meyer J, Sudbeck P, Held M, Wagner T, Schmitz ML, Bricarelli FD, Eggermont E, Friedrich U, Haas OA, Kobelt A, Leroy JG, van Maldergem L, Michel E, Mitulla B, Pfeiffer RA, Schinzel A, Schmidt H, Scherer G. Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Hum Mol Genet. 1997;6:91–8. [PubMed: 9002675]
- Ninomiya S, Narahara K, Tsuji K, Yokoyama Y, Ito S, Seino Y. Acampomelic campomelic syndrome and sex reversal associated with de novo t(12;17) translocation. Am J Med Genet. 1995;56:31–4. [PubMed: 7747782]
- Olney PN, Kean LS, Graham D, Elsas LJ, May KM. Campomelic syndrome and deletion of SOX9. Am J Med Genet. 1999;84:20–4. [PubMed: 10213041]
- Pfeifer D, Kist R, Dewar K, Devon K, Lander ES, Birren B, Korniszewski L, Back E, Scherer G. Campomelic dysplasia translocation breakpoints are scattered over 1 Mb proximal to SOX9: evidence for an extended control region. Am J Hum Genet. 1999;65:111–24. [PMC free article: PMC1378081] [PubMed: 10364523]
- Piper K, Ball SG, Keeling JW, Mansoor S, Wilson DI, Hanley NA. Novel SOX9 expression during human pancreas development correlates to abnormalities in campomelic dysplasia. Mech Dev. 2002;116:223–6. [PubMed: 12128229]
- Pop R, Conz C, Lindenberg KS, Blesson S, Schmalenberger B, Briault S, Pfeifer D, Scherer G. Screening of the 1 Mb SOX9 5' control region by array CGH identifies a large deletion in a case of campomelic dysplasia with XY sex reversal. J Med Genet. 2004;41:e47. [PMC free article: PMC1735745] [PubMed: 15060123]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Savarirayan R, Robertson SP, Bankier A, Rogers JG. Variable expression of campomelic dysplasia in a father and his 46,XY daughter. Pediatr Pathol Mol Med. 2003;22:37–46. [PubMed: 12687888]
- Schramm T, Gloning KP, Minderer S, Daumer-Haas C, Hörtnagel K, Nerlich A, Tutschek B. Prenatal sonographic diagnosis of skeletal dysplasias. Ultrasound Obstet Gynecol. 2009;34:160–70. [PubMed: 19548204]
- Smyk M, Obersztyn E, Nowakowska B, Bocian E, Cheung SW, Mazurczak T, Stankiewicz P. Recurrent SOX9 deletion campomelic dysplasia due to somatic mosaicism in the father. Am J Med Genet. 2007;143A:866–70. [PubMed: 17352389]
- Sock E, Pagon RA, Keymolen K, Lissens W, Wegner M, Scherer G. Loss of DNA-dependent dimerization of the transcription factor SOX9 as a cause for campomelic dysplasia. Hum Mol Genet. 2003;12:1439–47. [PubMed: 12783851]
- Staffler A, Hammel M, Wahlbuhl M, Bidlingmaier C, Flemmer AW, Pagel P, Nicolai T, Wegner M, Holzinger A. Heterozygous SOX9 mutations allowing for residual DNA-binding and transcriptional activation lead to the acampomelic variant of campomelic dysplasia. Hum Mutat. 2010;31:E1436–44. [PubMed: 20513132]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Thomas S, Winter R, Lonstein J. The treatment of progressive kyphoscoliosis in Camptomelic dysplasia. Spine. 1997;22:1330–7. [PubMed: 9201836]
- Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023;191:1164–209. [PMC free article: PMC10081954] [PubMed: 36779427]
- Velagaleti GV, Bien-Willner GA, Northup JK, Lockhart LH, Hawkins JC, Jalal SM, Withers M, Lupski JR, Stankiewicz P. Position effects due to chromosome breakpoints that map approximately 900 Kb upstream and approximately 1.3 Mb downstream of SOX9 in two individuals with campomelic dysplasia. Am J Hum Genet. 2005;76:652–62. [PMC free article: PMC1199302] [PubMed: 15726498]
- Vetro A, Ciccone R, Giorda R, Patricelli MG, Mina ED, Forlino A, Zuffardi O. XX males SRY negative: a confirmed cause of Infertility. J Med Genet. 2011;48:710–2. [PMC free article: PMC3178810] [PubMed: 21653197]
- Vidal VP, Chaboissier MC, de Rooij DG, Schedl A. Sox9 induces testis development in XX transgenic mice. Nat Genet. 2001;28:216–17. [PubMed: 11431689]
- von Bohlen AE, Böhm J, Pop R, Johnson DS, Tolmie J, Stücker R, Morris-Rosendahl D, Scherer G. A mutation creating an upstream initiation codon in the SOX9 5' UTR causes acampomelic campomelic dysplasia. Mol Genet Genomic Med. 2017;5:261–8. [PMC free article: PMC5441400] [PubMed: 28546996]
- Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, Pasantes J, Bricarelli FD, Keutel J, Hustert E, Wolf U, Tommerup N, Schempp W, Scherer G. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79:1111–20. [PubMed: 8001137]
- Walters-Sen LC, Thrush DL, Hickey SE, Hashimoto S, Reshmi S, Gastier-Foster JM, Pyatt RE, Astbury C. Atypical breakpoint in a t(6;17) translocation case of acampomelic campomelic dysplasia. Eur J Med Genet. 2014;57:315–8. [PubMed: 24821304]
- White S, Ohnesorg T, Notini A, Roeszler K, Hewitt J, Daggag H, Smith C, Turbitt E, Gustin S, van den Bergen J, Miles D, Western P, Arboleda V, Schumacher V, Gordon L, Bell K, Bengtsson H, Speed T, Hutson J, Warne G, Harley V, Koopman P, Vilain E, Sinclair A. Copy number variation in patients with disorders of sex development due to 46,XY gonadal dysgenesis. PLoS ONE. 2011;6:e17793. [PMC free article: PMC3049794] [PubMed: 21408189]
Publication Details
Author Information and Affiliations
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland
University of Freiburg
Freiburg, Germany
University of Lausanne;
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland
Publication History
Initial Posting: July 31, 2008; Last Revision: April 6, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Unger S, Scherer G, Superti-Furga A. Campomelic Dysplasia. 2008 Jul 31 [Updated 2023 Apr 6]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.