Clinical Description
Woodhouse-Sakati syndrome (WSS) is characterized by the endocrine findings of hypogonadism, diabetes mellitus, and hypothyroidism and progressive childhood-onset alopecia along with neurologic findings of progressive extrapyramidal movements, sensorineural hearing loss, and intellectual disability. To date, 88 individuals from more than 40 families have been reported [Steindl et al 2010, Nanda et al 2014, Abdulla et al 2015, Bohlega et al 2019].
Two clinical types of WSS have been described [Bohlega et al 2019] with variable prognosis. However, intrafamilial variability is common and both types can occur within a family:
Type 1. Severe and progressive neurologic disability at a younger age (range 9-17 years) causing significant impairment of the quality of life and activities of daily living. Manifestations include severe intellectual disability and dystonia.
Type 2. Absent or mild neurologic involvements that do not affect activities of daily living
Endocrine
Hypogonadism, present in all affected individuals, manifests as delayed puberty with lack of secondary sexual characteristics. The nature of hypogonadism has been difficult to characterize as both hypergonadotropic and hypogonadotropic hypogonadism have been described [Agopiantz et al 2014]; in about 30% of affected individuals the hormonal profile does not neatly fit either group. Sense of smell is normal.
Women typically have primary amenorrhea. Detailed endocrine investigation in more than 50 of the women described in the literature typically revealed severely reduced or absent estradiol and high FSH and LH, consistent with hypergonadotropic hypogonadism. There appears to be decreased hypothalamic-pituitary responsiveness, as the FSH and LH are not as high as expected for the degree of ovarian failure [Woodhouse & Sakati 1983, Rachmiel et al 2011, Agopiantz et al 2014].
The ovaries are streak or underdeveloped, and not visualized by laparotomy, laparoscopy, or autopsy [Al-Semari & Bohlega 2007, Ben-Omran et al 2011, Rachmiel et al 2011]. Ovarian biopsy showed fibrous tissue with no identifiable oocysts [Woodhouse & Sakati 1983, Agopiantz et al 2014].
Men have moderately low testosterone and – in contrast to women – inappropriately low gonadotropins, consistent with hypogonadotropic hypogonadism, which may be of central or central and peripheral etiology. Semen analysis may show azoospermia [Agopiantz et al 2014, Ali et al 2016]. One male had cryptorchidism [Rachmiel et al 2011].
Although the testes are of normal size, testicular biopsy reveals reduced spermatogenesis with predominance of Sertoli cells and few Leydig cells [Agopiantz et al 2014].
Low insulin-like growth factor 1 (IGF-1) is present in all individuals [Ali et al 2016]. Reduction of IGF-1 is more pronounced in females [Al-Semari & Bohlega 2007, Ben-Omran et al 2011]. The low IGF-1 levels may reflect low sex steroids resulting from hypogonadism.
The growth pattern is normal and growth hormone levels are usually normal; short stature is not a part of this syndrome [Agopiantz et al 2014].
Diabetes mellitus. Type 2 diabetes (either insulin dependent or non-insulin dependent) was reported in 66% of all individuals and 96% of those older than age 25 years [Al-Semari & Bohlega 2007, Agopiantz et al 2014].
Hypothyroidism of peripheral origin (primary but without evidence of autoimmunity) was found in 30% of individuals, typically around age 20 years [Al-Semari & Bohlega 2007].
Other. No abnormalities of the corticotropic axis or prolactin [Al-Semari & Bohlega 2007, Agopiantz et al 2014] have been reported.
Ectodermal
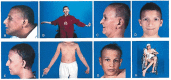
Alopecia. All affected individuals have predominantly frontotemporal alopecia with sparse, thin scalp hair. Hair loss begins in childhood and often progresses to alopecia totalis in the third or fourth decade or earlier. Eyelashes and eyebrows are absent or sparse. In men, facial hair is absent or underdeveloped.
Scanning electron microscopy of the hair shows longitudinal grooves with no specific abnormalities [Al-Semari & Bohlega 2007].
Facial skin is often wrinkled in advanced stages, conferring a progeroid appearance [Woodhouse & Sakati 1983, Al-Semari & Bohlega 2007, Agopiantz et al 2014].
Anodontia. Total loss of teeth is rare; when it occurs, it is usually seen at a later stage [Al-Semari & Bohlega 2007, Agopiantz et al 2014].
Nails appear to be normal.
Neurologic
Extrapyramidal abnormal movement was seen in more than 56% of reported individuals. In particular, dystonic spasms with dystonic posturing were seen in the majority, including segmental dystonia affecting the craniocervical region, oromandibular region, or one extremity. Often, the first neurologic manifestation is abnormal posturing movements that typically start insidiously in childhood or the early teens. Dysarthria (often with a high-pitched voice) and dysphagia are common.
In a majority of individuals, dystonia becomes generalized and disabling (, , ). Progressive dystonia of the trunk may lead to severe dystonic scoliosis. As the dystonia progresses, gait difficulties ultimately lead to immobility.
Inter- and intrafamilial variability is common [Al-Semari & Bohlega 2007, Ben-Omran et al 2011, Ali et al 2016 , Bohlega et al 2019]. For example, families with the founder DCAF17 pathogenic variant (c.436delC) can have dystonia [Al-Semari & Bohlega 2007] or not [Bohlega et al 2019]. Of note, although extrapyramidal features were not mentioned in the original report of Woodhouse and Sakati [1983], it was found that up to 65% of affected individuals develop variable degrees of dystonia at some point in the disease course () [Bohlega et al 2019 ].
Sensorineural hearing loss (SNHL). Moderate bilateral SNHL was noted in 62% of reports. When present, deafness is invariably postlingual, usually starting in adolescence [Ben-Omran et al 2011].
Intellectual disability is described in 58% of individuals. It is typically mild and usually overshadowed by accompanying severe and disabling dystonia, dysarthria, and SNHL. The authors have observed ten individuals who were able to complete a college education and hold permanent manual occupations [Author, personal observation].
Other. Seizures with onset in early childhood, tremors, and mild Parkinsonism features have rarely been reported [Al-Semari & Bohlega 2007, Schneider & Bhatia 2008, Bohlega et al 2019].
Polyneuropathy with stocking glove sensory loss and diminished deep tendon reflexes but normal strength has been reported [Schneider & Bhatia 2008].
Other Findings
Dysmorphic facial features include a long triangular face, prominent nasal bridge, widely spaced eyes, and sparse eyebrows, creating a characteristic facial appearance (, , ).
Bilateral keratoconus was reported in four individuals [Al-Swailem et al 2006, Schneider & Bhatia 2008, Ben-Omran et al 2011].
Electrocardiographic (EKG) abnormalities (lengthening of the ST segments and T-wave flattening) were described in the original report [Woodhouse & Sakati 1983] but rarely reported subsequently [Koshy et al 2008, Schneider & Bhatia 2008]. Of note, these EKG abnormalities were asymptomatic and individuals with WSS have no major cardiac manifestations.