Clinical Description
The TP63-related disorders include the overlapping phenotypes summarized in Table 2 and fully described in the text that follows.
AEC Syndrome
The manifestations of ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome are typically present at birth.
Ankyloblepharon is present in 70% of neonates. While the upper- and lower-eyelid adhesions can be obvious, partial adhesion of the upper and lower eyelids can be subtle and the filiform adhesions can spontaneously lyse before they are recognized as such.
Lacrimal puncta are frequently absent, often leading to chronic conjunctivitis and blepharitis, which is often not recognized in infancy but seen in early childhood [Sutton et al 2009].
Ectodermal defects
Skin. Nearly 100% of affected neonates have superficial skin erosions that vary from very limited to severe, life-threatening full-body involvement. The erosions most typically affect the scalp at birth and during infancy. Severe scalp erosions often lead to scarring alopecia and hypotrichosis. This is NOT seen in other TP63-related disorders.
The skin erosions tend to be recurrent and intermittent throughout childhood and into adulthood with frequent involvement of the head and neck, palms, soles, and skin folds.
Congenital erythroderma (i.e., diffuse erythema with associated erosions) is observed in 70%-90% of infants. The skin can also appear shiny with a collodion membrane (red, shiny, membranous skin changes) [
Siegfried et al 2005].
Children typically manifest cutaneous depigmentation and scarring, most likely due to postinflammatory pigmentary changes related to previous erythroderma and associated underlying erosions that may or may not be appreciated clinically. African American infants can have facial hypopigmentation in a mask pattern that improves with age. Affected individuals with fair skin typically have a reticulated hyperpigmentation on the neck and intertriginous areas that progresses with age to cribriform, reticulate, stellate, or punctate scarring most commonly on the shoulders, upper back, and chest.
Histopathologic features of skin biopsies may reveal epidermal atrophy, pigment incontinence, and a prominent superficial perivascular plexus with limited lymphocytic infiltrate [
Dishop et al 2009].
Hair changes become more obvious with age. Hair is typically light colored and coarse, wiry, and brittle with a spun-glass/gold or "uncombable" appearance. Eyebrows and eyelashes are sparse. Light and scanning microscopy may reveal structural and pigmentary alterations of the hair including kinking, grooves, and discontinuous pigmentation.
Nail
changes, present in all and more obvious with age, vary among individuals. Most affected individuals have nail dystrophy (abnormal nail plate texture) and hyperconvex nail plates. Micronychia (abnormally small nail plates), distal frayed edges with nail plate resorption, and absent nails are also frequent [
Julapalli et al 2009].
Dental anomalies. Malformed teeth (conical shape with small occlusal tables) and hypodontia (reduced number of teeth) also become evident during childhood and adolescence. Affected adults have an average of 4.75 secondary teeth [
Farrington & Lausten 2009].
Clefting is present in all. Clefting can include submucous cleft palate only, cleft of the soft and/or the hard palate only, cleft lip only, or the combination of cleft lip and cleft palate [Cole et al 2009].
Other findings include the following:
Limb anomalies were initially not considered to be part of the syndrome, but syndactyly of fingers and toes and/or camptodactyly (permanent and irreducible flexion of the fingers) of hands have been seen. Split-hand/foot malformation was observed in two of 17 individuals (12%) with AEC syndrome [
Sutton et al 2009].
Facial features become more distinctive with age. Findings commonly include maxillary hypoplasia, micrognathia, broad nasal root, underdeveloped alae nasi, thin vermilion of the upper lip, and short philtrum.
Trismus has been reported in 35% of individuals with AEC syndrome [
Sutton et al 2009].
Hearing loss. More than 90% of children have conductive hearing loss, often with secondary speech delay [
Cole et al 2009].
Growth. Poor weight gain and failure to thrive should be anticipated. When treated appropriately with nutritional supplementation poor weight gain improves with age.
Linear growth abnormalities are observed in early childhood with a significantly lower height for age compared to the reference population. The growth pattern in AEC is similar to that reported for hypohidrotic ectodermal dysplasia [
Motil & Fete 2009].
Psychological impact related to the phenotypic features of the disease can include a reduced quality of life with negative impact on both child and family. In one study, a variable degree of psychological functioning was noted with some families reporting few ill effects from the disease while others reported significant impact [
Lane et al 2009].
ADULT Syndrome
The manifestations of acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome are typically present at birth (although they may become more prominent with age), with the exception of skin freckling.
Limb anomalies. Syndactyly of fingers and toes is most commonly seen.
Ectodermal defects
Skin tends to be dry but erosions are not present.
Hair changes are more obvious with age. Hair is typically light colored and fine. Eyebrows and eyelashes are sparse.
Nail dysplasia is commonly reported.
Dental anomalies. Malformed teeth (conical shape with small occlusal tables) and hypodontia (reduced number of teeth) also become evident during childhood and adolescence.
Sweating. Subjective decreased sweat production is universal and is reported as heat intolerance; however, this does NOT lead to hyperthermia or fevers as seen in
hypohidrotic ectodermal dysplasia.
Lacrimal duct atresia is frequent and often leads to chronic conjunctivitis and blepharitis, which are often not recognized until early childhood [Sutton et al 2009].
Breast and/or nipple hypoplasia is seen commonly and most notably in females. This feature is characteristic of ADULT and limb-mammary syndrome and NOT typically seen in other TP63-related disorders.
Excessive freckling in sun-exposed areas is seen in a subset of affected individuals and progresses with age and sun exposure. This feature is NOT seen in other TP63-related disorders.
EEC3
The manifestations of ectrodactyly, ectodermal dysplasia, cleft lip/palate syndrome 3 (EEC3) are typically present at birth.
Limb anomalies are seen in 68%-90% of individuals with 60% having tetramelic involvement. A wide variety of limb abnormalities are reported including syndactyly, oligodactyly, split-hand/foot malformation, and digital duplication. A cohort of 152 individuals with EEC syndrome showed split-hand/foot malformation in 68% and syndactyly in 43% [Rinne et al 2006a].
Ectodermal defects
Skin tends to be dry but erosions are not present.
Hair changes become more obvious with age and are seen in 60%-80% of individuals with EEC syndrome [
Rinne et al 2006a]. Hair is typically silvery blond, coarse, and dry; 20% have sparse hair. Light microscopy has been reported to be normal in EEC syndrome [
Pashayan et al 1974]. Eyebrows and eyelashes are sparse.
Nail
dysplasia is commonly reported.
Dental anomalies. Malformed teeth (conical shape with small occlusal tables) and hypodontia (reduced number of teeth) also become evident during childhood and adolescence.
Cleft lip with or without cleft palate is present in 60%-75% and is bilateral in half of cases. Clefting can include submucous cleft palate only, cleft of the soft and/or the hard palate only, cleft lip only, or the combination of cleft lip and cleft palate [Buss et al 1995].
Absent lacrimal puncta is reported in 90% of individuals and results in tearing, blepharitis, dacryocystitis, keratoconjunctivitis, and photophobia and often leads to corneal ulceration and scarring [Buss et al 1995].
Genitourinary malformations are reported in 45% and may include hypospadias and developmental abnormalities of the kidneys and urinary collecting system.
Limb-Mammary Syndrome
The manifestations of limb-mammary syndrome are typically present at birth.
Limb anomalies including split-hand/foot malformation and syndactyly are reported in 75%-85% of individuals.
Breast and/or nipple hypoplasia is seen commonly with almost all individuals having nipple aplasia or hypoplasia and 90% of females having mammary gland aplasia or hypoplasia. This feature is characteristic of ADULT and limb-mammary syndrome and NOT typically seen in other TP63-related disorders.
Lacrimal duct atresia is seen in about half leading to chronic conjunctivitis and blepharitis, which are often not recognized until early childhood [van Bokhoven et al 1999].
Ectodermal defects
Note: Skin and hair abnormalities are NOT typically seen, in contrast to other TP63-related disorders.
Cleft lip with or without cleft palate is present in 25%-30% of individuals and can include submucous cleft palate only, cleft of the soft and/or the hard palate only, cleft lip only, or the combination of cleft lip and cleft palate.
SHFM4
The manifestations of split-hand/foot malformation-4 (SHFM4) are typically present at birth.
Limb abnormalities include median clefts of the hands and feet; aplasia/hypoplasia of phalanges, metacarpals, and metatarsals; and some syndactyly.
Ectodermal abnormalities and cleft lip/palate are considered to be exclusion criteria for making the diagnosis SHFM4.
Isolated Cleft Lip/Palate (Orofacial Cleft 8)
Identification of a TP63 pathogenic variant in three individuals ‒ a girl age 4 years; a boy age 3 years and his father; all with apparent nonsyndromic cleft lip/palate ‒ have been reported by Leoyklang et al [2006] and Basha et al [2018], respectively. Further evaluations revealed no other features of TP63-related disorders in these individuals.
Khandelwal et al [2019] identified partial deletions of TP63 in individuals from three families with orofacial cleft and/or hypodontia and minor anomalies of the skin and nails.
Genotype-Phenotype Correlations
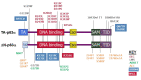
Note: Pathogenic variants have been described on two TP63 isoforms: the TAp63α isoform, encoded by NM_003722.4, and the ΔNp63α isoform, encoded by NM_001114982.1, which is 39 amino acids shorter and has an alternate N-terminal TA domain. See for details.
Typical and common TP63 pathogenic variants identified in various disorders as indicated by color key. Pathogenic variants indicated by * are specific for the ΔNp63α isoform and their numbering is based on the respective reference sequences (more...)
AEC syndrome. All pathogenic variants associated with AEC syndrome occur in either the sterile alpha motif (SAM) domain (82%) or the ΔNp63-specific N-terminal domain (18%). Pathogenic variants in the N-terminal domain that introduce premature termination codons lead to the use of an alternative start codon [Rinne et al 2008] and to the consequent production of ΔNp63α isoforms lacking the N-terminal domain, which are specifically associated with AEC syndrome. This isoform of p63 is the predominant isoform in mature epidermis, and it has been shown to repress ZNF750, leading to impaired epidermal differentiation [Zarnegar et al 2012].
ADULT syndrome is typically associated with pathogenic variants in the DNA binding domain. Other pathogenic variants have been reported in isolated individuals with features reminiscent of ADULT syndrome, but also of other TP63-associated syndromes in ΔNp63α (an alternative TA domain), in TAp63α between the TA and DNA binding domains [Rinne et al 2006b, Rinne et al 2007], and at other locations in TAp63α [van Zelst-Stams & van Steensel 2009].
EEC3. All EEC3-causing pathogenic variants are missense variants in the DNA binding domain and have been demonstrated to disrupt DNA binding [Rinne et al 2006b]. Splice changes and frameshifts associated with EEC3 have been reported [Celli et al 1999, van Bokhoven et al 2001, Barrow et al 2002, Monti et al 2013].
Limb-mammary syndrome is caused by pathogenic missense variants that are located between the transactivation domain and the DNA binding domain (p.Gly115, p.Ser129, and p.Gly173 residues in TAp63α) or by truncating variants in the SAM domain of TP63 [van Bokhoven et al 2001, Rinne et al 2007].
SHFM4. Pathogenic missense variants in the TA and DNA binding domains have been associated with SHFM4 [Rinne et al 2007].
Orofacial cleft 8 has been associated with a TP63 variant in the DNA binding domain [Leoyklang et al 2006, Basha et al 2018].
Nomenclature
Ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome is also known as Hay-Wells syndrome, after the physicians who first described the condition in 1976.
Rapp-Hodgkin syndrome (RHS), once considered a separate entity, is now considered to be part of the spectrum of the AEC syndrome because of the overlap of clinical manifestations and TP63 pathogenic variants in the two conditions [Cambiaghi et al 1994, McGrath et al 2001].
EEC3 is thought to be genetically unrelated to EEC1 (which has been mapped to chromosome 7q11q21). An entity called EEC2 was initially mapped to chromosome 19 [O'Quinn et al 1998]; however, pathogenic variants in TP63 were ultimately identified [Celli et al 1999].