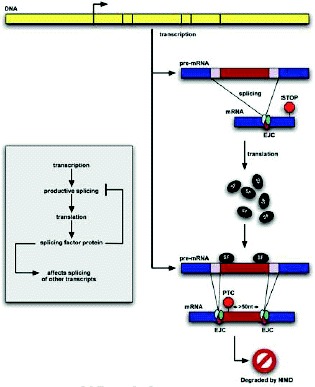
Most human genes exhibit alternative splicing, but not all alternatively spliced transcripts produce functional proteins. Computational and experimental results indicate that roughly a third of reliably inferred alternative splicing events in humans result in mRNA isoforms that harbor a premature termination codon (PTC). These transcripts are predicted to be degraded by the NMD pathway. One potential explanation for this startling observation is that cells routinely link alternative splicing and NMD to regulate the abundance of mRNA transcripts. This mechanism, which we call “Regulated Unproductive Splicing and Translation” (RUST), has been experimentally shown to regulate the expression of a wide variety of genes in many organisms from yeast to humans. It is frequently employed to autoregulate proteins that affect the splicing process itself. Thus, alternative splicing and NMD, acting together, play an important and widespread role in regulating gene expression.
Introduction
One major result of the large-scale sequencing projects of the last decade has been an appreciation of the extent of alternative splicing of mammalian transcripts. Estimates vary, but most reports agree that over half of human genes generate transcripts that are alternatively spliced.1,2 What is the biological function of this extensive alternative splicing? Many claim it is a mechanism of proteome expansion,3 but relatively few alternative forms encode truly distinct proteins. More often, alternative splicing seems to modulate gene function by adding or removing protein domains, affecting protein activity, or altering the stability of the transcript or the resulting protein.4-6
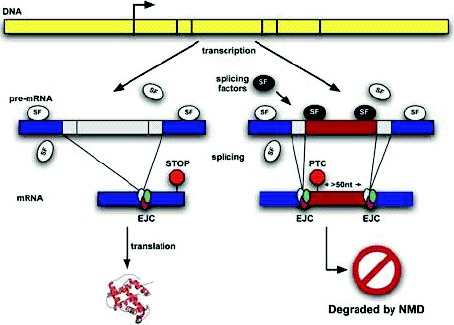
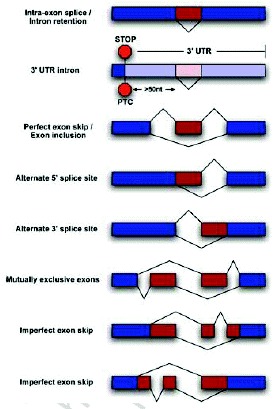
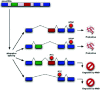
In the last few years, it has become clear that many alternative splice forms previously thought to encode truncated proteins are actually targets of NMD (Fig. 1). In mammals, a termination codon located more than about 50 nucleotides upstream of an exon-exon junction is generally recognized as premature, eliciting NMD7,8 (see also chapter by Maquat). An understanding of this rule allowed identification of numerous transcripts that are predicted to be degraded rather than translated into protein. Such transcripts can arise through various patterns of alternative splicing (Fig. 2), which may introduce an in-frame termination codon, may induce a frameshift which gives rise to a downstream termination codon, or may introduce an exon-exon junction downstream of the original stop codon. The prevalence of these NMD-targeted transcripts calls for a reconsideration of the roles of alternative splicing and NMD.
NMD was originally considered to be a quality control mechanism, protecting cells from the potentially toxic effects of nonsense mutations, errors in transcription, and errors in splicing.9,10 We now know that there are many natural targets of NMD (see chapters by He and Jacobson, and Sharifi and Dietz), including transcripts with upstream open translational reading frames (uORFs), products of alternative splicing, byproducts of V(D)J recombination, and transcripts arising from transposons and retroviruses.11 Indeed, it now seems that a major effect of NMD is to downregulate physiological transcripts, in addition to clearing cells of erroneous transcripts.
Alternative Splice Forms Are Frequently Targets of NMD
While it was long known that alternative splicing may produce isoforms that are degraded by NMD, this was not appreciated as a pervasive process until 2002. At that point, genome-wide studies provided the first indication that a substantial fraction of human genes are routinely spliced to produce isoforms that are targeted for NMD.
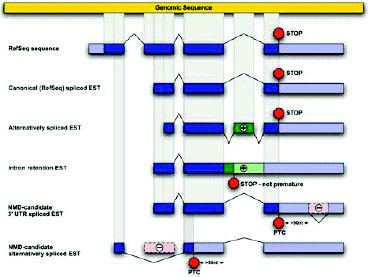
In the first study to show widespread predicted NMD of alternative splice forms, Lewis et al used human mRNA and expressed sequence tags (ESTs) available from public databases to infer alternative splice forms and identify PTCs.12 Although the data set considered was limited, it revealed that at least 12% of genes for which RefSeq mRNA sequences are available generate a PTC+ isoform. The actual prevalence of such genes may be substantially higher than this lower bound. This study considered 16,780 human mRNA sequences from the reviewed category of RefSeq,13 a set of well-characterized, experimentally confirmed transcript sequences. Alignment of the RefSeq mRNAs to their genomic loci showed that 617 of these curated mRNA sequences, or 3.7%, contained PTCs. The alternative splice forms inferred by aligning EST sequences from dbEST14 to the remaining 16,163 genomic loci (Fig. 3) substantially increased the estimated fraction of genes with PTC+ isoforms. Based on the EST data, over 3000 of the RefSeq genes had alternative splice forms, and 45% of these alternatively spliced genes had at least one form that was predicted to be a target of NMD.12

These results have been confirmed and strengthened by later studies. An analysis of the isoforms contained in SWISS-PROT15 showed that even this reliable, curated database contained presumed translation products of mRNA sequences that were more likely to have been degraded by NMD. Alignment of the mRNA sequence of each protein isoform reported in SWISS-PROT to the human genome identified reliable exon-intron structures for 2483 isoforms from 1363 genes. The 50-nucleotide rule predicts that 144 isoforms (5.8% of 2483) from 107 genes (7.9% of 1363) contain a PTC of the type that should elicit NMD.16
A new study by Baek and Green has extended the analysis of PTC+ alternative splicing to consider conservation of splice forms between human and mouse.17 This approach helps distinguish aberrant splice events from rare but functional variants. Starting from a large set of cDNA and EST sequences, Baek and Green identified about 1500 pairs of exon inclusion/exclusion splice forms found in both human and mouse. A quarter of the conserved alternative forms contain a conserved PTC,17 suggesting that these isoforms play a functional role, and that the PTC is important to their function.
Direct experimental evidence from human cells supports these computational results. Mendell and coworkers made HeLa cells depleted of Upf1, an essential component of the NMD pathway, and used microarrays to compare the abundance of mRNAs in these cells to the abundance of mRNAs in unmodified cells.11 They found that 4.9% of the ˜4000 transcripts tested showed significantly higher abundance in cells deficient in NMD, suggesting that NMD normally downregulates those transcripts. They confirmed that their observations were largely due to the direct action of NMD, rather than being a downstream regulatory consequence, by showing that several of the putative NMD-targeted transcripts decayed faster in normal cells than in cells depleted of Upf1. They also confirmed that the effect they observed was due to NMD and not to the action of Upf1 in some other pathway, by showing that the abundance of PTC+ transcripts was similarly upregulated upon the depletion of Upf2, another protein essential for NMD. Finally, Mendell et al also observed that 4.3% of transcripts were decreased in abundance in NMD-deficient cells. The stability of those transcripts was not altered by NMD deficiency, showing that the change in their abundance was an indirect effect.
The striking prevalence of PTC+ alternative splice forms begs for an explanation. Why would cells routinely expend energy and resources transcribing, splicing, and degrading PTC+ isoforms? Are these isoforms often translated after all, contravening the 50-nucleotide rule? Are the observed PTC+ isoforms all due to transcriptional or splicing noise? Are they an unavoidable side effect of productive alternative splicing, which itself is conserved as an important mechanism for producing a diversity of proteins? Or, does the combination of alternative splicing and NMD constitute a novel system for regulating gene expression? We will consider each of these potential explanations in turn.
Do the Observed PTC+ mRNA Isoforms Evade NMD to Produce Functional Protein?
The existence of numerous PTC+ isoforms was first inferred from EST data.12 One may wonder why EST evidence exists at all for isoforms that are expected to be degraded by NMD. As observed in numerous experiments (Table 1), NMD substantially reduces the abundance of PTC+ transcripts, but it does not eliminate the transcripts entirely. One explanation is that NMD surveillance may not be completely effective. Furthermore, PTC+ isoforms are not degraded instantly upon being spliced; rather, their degradation occurs only as a consequence of a pioneer round of translation,18 which for most mRNAs might occur near the nuclear pore during or soon after export of the mRNA from the nucleus (reviewed in ref. 19). Thus, we expect some steady-state abundance of PTC+ isoforms that have not yet been degraded, especially inside the nucleus, or that have escaped decay, particularly in the cytoplasm. A series of elegant experiments and computational modeling in yeast20 suggests that the dominant reason for the presence of PTC+ mRNAs in the yeast cell is the temporal lag between splicing and degradation, rather than incomplete surveillance. A similar temporal lag may occur in mammals, despite differences in NMD dynamics between mammals and yeast. Evidently, the resulting abundance of PTC+ isoforms is in many cases high enough for ESTs deriving from those isoforms to be observed and deposited in dbEST. Indeed, many of the alternative splice junctions that generate a PTC are supported by two or more ESTs.
Table 1
Experimentally confirmed examples of unproductive splicing.
EST libraries are nonetheless biased against less stable isoforms. Using sequence features such as splice-site strength, Baek and Green modeled the predicted inclusion rates of alternative exons.17 They showed that PTC+ isoforms are probably produced at a higher rate than they are observed in EST data, but are degraded before they can be sequenced. Thus, the EST data underestimate the fraction of PTC+ mRNA deriving from a given gene, and also underestimate the number of genes with PTC+ alternative splicing. For this reason, and also because the quality filters used in the above studies excluded many genes and isoforms, reports offer a lower bound on the number of PTC+ isoforms; the true prevalence of alternative splicing and of PTC+ isoforms may be substantially higher.
Some PTC+ transcripts may evade NMD, increasing their likelihood of being observed and deposited in sequence databases. There are a few known examples in which a transcript that should be degraded according to the 50-nucleotide rule is in fact stable and translated to make protein. These include polycistronic transcripts on which translation is reinitiated downstream of a PTC;21-23 apolipoprotein B, which is protected from NMD by an RNA editing complex;24 some transcripts with a PTC near the initiation codon;25 and an aberrant beta-globin transcript which is protected from NMD by an unknown mechanism.26 Although NMD does not prevent protein production entirely in such cases, it may nonetheless limit expression from PTC+ transcripts substantially, as was shown for an alternative transcript of fumarylacetoacetate hydrolase (FAH) and for activator of RNA decay (ARD-1).21,27
Nonetheless, documented exceptions to the 50-nucleotide rule are rare, and there are many more known cases in which the rule is honored; indeed, the microarray results described above are consistent with the rule, as are diverse experiments on individual transcripts (Table 1).
Even for transcripts that are degraded by NMD, the possibility remains that the truncated protein products of the pioneer round of translation are functionally significant, since some regulatory proteins can have an effect even at very low copy number.28 Also, to the extent that NMD is not completely effective at detecting and degrading PTC+ transcripts, the overlooked transcripts may be translated to produce truncated proteins. However, these proteins will frequently lack critical domains, rendering them inactive or even harmful. In any case, it is hard to imagine that functional roles of truncated proteins could explain the high prevalence of genes with PTC+ isoforms, especially given the wide functional diversity of those genes, and no data exist to support such a view.
While there may be exceptions, it seems unlikely that many PTC+ isoforms produce functional protein due to incomplete surveillance or by otherwise evading NMD.
Do the Observed PTC+ mRNA Isoforms Represent Missplicing or Cellular Noise?
NMD was originally described as a means of clearing erroneous transcripts from the cell.9,10 In keeping with this role, some alternative splice forms that are degraded by NMD could represent splicing errors. Such errors could arise from mutations disrupting splice sites or regulatory sequences, including mutations in intronic regions that are invisible after intron removal. Also, the splicing machinery itself could recognize incorrect splice sites. The spliceosome distinguishes true splice sites from nearby cryptic sites with impressive fidelity, but splice site recognition is a complex process and errors must occur at some low rate. There are at present no clear data on the extent of missplicing. However, since EST libraries contain millions of transcript sequences, even extremely rare events may be represented.
In EST-based computational analyses, splicing errors can be distinguished from genuine alternative splicing to some extent by filtering out splicing events that are seen only in a few ESTs. However, filtering will certainly exclude some legitimate rare splice forms as well. With multiple mammalian genomes available, recent work has focused on evolutionary conservation to suggest positive selection and, perhaps, functional roles for conserved alternative forms.29
Conservation of alternative splice forms between closely related organisms can be used to distinguish functional alternative splicing from probable splicing errors. Minor isoforms, i.e., those that occur only a fraction of the time, are less often conserved than major isoforms,30 and they may sometimes represent recent mutations or splicing errors. Those minor isoforms that are conserved, including PTC+ isoforms, are more likely to be functional than minor isoforms that are seen only in one species.31
As described above, Baek and Green identified PTC+ alternative splice forms that were conserved between human and mouse, to filter out aberrant splicing. They noted that the inclusion of the same “accidental” alternative exon is unlikely to happen by chance in both species, because accidental recognition of the same position in two species—a position that is not under selective pressure to be recognized as a splice site—is unlikely. On the other hand, occasional accidental skipping of the same exon could easily be seen in both human and mouse; if the spliceosome were to miss bone fide splice sites at some low frequency, the same accident might be found by chance in homologous transcripts in two species. To reduce the influence of these conserved but aberrant splices on their data set, they designed a statistical method to discriminate between splicing errors and functional alternative splicing. Using this method, 80% of the conserved PTC-producing splice events were legitimate, compared to 20% that appeared aberrant.17 Thus, most of the conserved PTC-producing splice events were not due to missplicing.
Are the Observed PTC+ mRNA Isoforms a Side Effect of Productive Alternative Splicing?
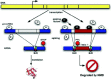
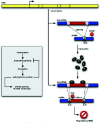
In the particular situation of mutually exclusive exon usage, NMD may be a mechanism for removing transcripts that erroneously include both exons or neither exon. In some instances there are physical constraints that force the splicing machinery to include one exon or the other but not both.32 In other cases, including both exons or neither exon would introduce a frameshift resulting in a PTC, targeting the mRNA for degradation. In the case of fibroblast growth factor receptor (FGFR)2 RNA, an isoform including exon IIIb while skipping exon IIIc is productive; similarly, the isoform including exon IIIc but excluding exon IIIb is productive. However, the spliceosome may instead pair the same splice sites differently such that both exons are included, or such that neither is included. Each of these latter possibilities introduces a PTC (Fig. 4).
Each of the splice sites involved in the removal of exons IIIb and IIIc is required for the production of at least one productive isoform; the unproductive isoforms arise simply from alternate pairings of these otherwise productive splice sites. Given that the spliceosome is prone to such alternate pairings, there may be evolutionary pressure to ensure that the undesired isoforms include a PTC. This case differs from the quality control scenario described above, in that the degraded isoforms result not from random noise but as an inevitable side effect of the mechanism for productive alternative splicing. NMD is used as a filter to remove these “side-effect” isoforms, which may comprise a substantial fraction of the transcripts produced (up to 50% in the case of FGFR2 RNA).33
We examined the alternative isoforms inferred from human dbEST data (above), and found that PTC+ isoforms could be explained as this kind of side effect for 34% of the genes that produce them. That is, 66% of the genes that generate a PTC+ isoform have a splice site that is specific to PTC+ isoforms and that is responsible for introducing the PTC (D.A.W. Soergel, unpublished data). If these unproductive isoforms were on the whole detrimental to the cell, then we would expect evolution to have eliminated the PTC+-specific splice sites long ago; but in fact many of them are strikingly conserved, as we discuss below. Thus, while the contribution of “side-effect” isoforms may be significant, they alone cannot explain the high prevalence of PTC+ isoforms.
Are the Observed PTC+ mRNA Isoforms Part of a Mechanism for Regulating Gene Expression?
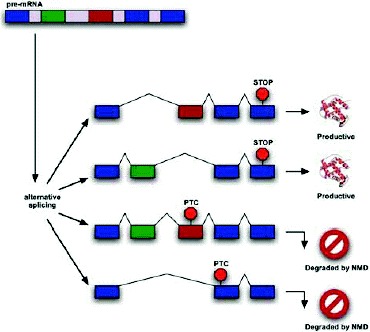
None of the phenomena discussed above—NMD evasion, noise, and “side-effect” splicing—are sufficient to explain the high prevalence of PTC+ isoforms that are observed. A remaining explanation is that the cell commonly produces a substantial fraction of NMD-targeted isoforms in a functional or regulated manner. This process may provide an additional level of regulatory circuitry to help the cell achieve the proper level of expression for a given protein. The cell could change the level of productive mRNA after transcription by shunting some fraction of the already-transcribed pre-mRNA into an unproductive splice form and thence to the decay pathway (Fig. 1).
The literature contains numerous examples in which a regulatory process involving alternative splicing and NMD has been experimentally confirmed (Table 1), and many more examples in which experiments are consistent with and suggestive of this mode of regulation (Table 2). We propose that gene regulation through the coupled action of alternative splicing and NMD is widespread, and that this is a major explanation for the large number of observed PTC+ isoforms. We have termed this process “regulated unproductive splicing and translation,” or RUST.
Table 2
Putative examples of unproductive splicing.
RUST can be used to regulate protein levels, and the process is itself regulated by changes in the splicing environment. In the simplest case, some constant fraction of pre-mRNA transcribed from a given gene is spliced into an unproductive, NMD-targeted form. In other cases, the proportion of transcripts targeted for degradation is regulated by an external input. Finally, autoregulatory loops can arise in which a protein affects the splicing pattern of its own pre-mRNA.
Constitutive Unproductive Splicing
The simplest type of coupled alternative splicing and NMD is one in which the ratio of productive to unproductive splice forms is not significantly variable. In this case, the combined effect of alternative splicing with NMD reduces mRNA abundance by a more or less constant factor. An apparent example of this is provided by the Calpain-10 transcript, which encodes a ubiquitously expressed protease. This transcript is alternatively spliced to produce eight mRNA isoforms.16,34 Of these, our analysis of SWISS-PROT and genomic sequences showed that four contain PTCs. An expression study by Horikawa et al showed that these very isoforms were “less abundant” in vivo than the other four.34 An experiment in our lab later showed that the PTC+ isoforms increased in abundance relative to the PTC- isoforms when cells were treated with cycloheximide, which blocks translation and thereby inactivates the NMD pathway.35 This result is consistent with the idea that all eight mRNA isoforms are produced but that the four PTC+ isoforms are degraded by NMD. Numerous transcripts in the literature are similarly processed (Table 1a). Of course, in each case, there may be as-yet-unknown regulatory inputs to splicing that do alter the isoform proportions.
Regulated Unproductive Splicing
Many examples of regulated alternative splicing leading to NMD are also known (Table 1b). In addition to changing the relative abundance of different functions, changes in the splicing environment may increase or decrease the production of functional isoforms relative to PTC+ isoforms that are degraded by NMD (Fig. 1).
The spliceosome recognizes a range of related sequence signals as 5' and 3' splice sites, with a range of “strengths” or binding affinities. Selection of splice sites is also under the control of a host of regulatory splicing factors, which bind to specific sequence signals on the pre-mRNA. These sequences may be exonic or intronic, and may be associated with enhancement or suppression of splicing at nearby (and sometimes at distant) splice sites. Exonic splicing enhancers (ESEs), intronic splicing enhancers (ISEs), exonic splicing silencers (ESSs), and intronic splicing silencers (ISSs) are frequently found in clusters, suggesting combinatorial regulation of splicing by complexes of splicing factors.36,37
Through the selection of alternative splice sites, splicing can give rise to a PTC in various ways. Inclusion of an alternative exon can introduce a PTC directly or shift the frame of the downstream exons to cause a downstream PTC. Similarly, exclusion of an exon can result in a frameshift and PTC. Finally, removal of an intron from the 3' untranslated region (UTR) may cause the normal stop codon to trigger NMD.
A change in the abundance of splicing factors can shift the balance of splicing patterns towards the production of NMD-targeted isoforms, thereby reducing the abundance of productive transcripts and hence the rate of protein production. In this way, splicing factors can alter gene expression, analogous to transcription factors.
This intriguing mechanism is used to regulate expression of MID1, a microtubule-associated protein involved in triggering degradation of phosphatase 2A.38 This gene is ubiquitously transcribed, but its transcripts are spliced in a tissue- and development-specific manner. Winter and coworkers observed numerous alternatively spliced transcripts that included alternative exons, in addition to the nine previously known exons. Most of these novel exons contained in-frame stop codons. Some of these stop codons were followed by alternative poly(A) tails, allowing translation of a C-terminally truncated protein. A second class of alternative transcripts contained stop codons closely followed by an in-frame start codon, suggesting the possibility of translation reinitiation and production of N-terminally truncated protein. A third class of alternative transcripts contained PTCs that were associated neither with an alternate poly(A) signal nor with an alternate translation start site. These transcripts should be subject to NMD according to the 50-nucleotide rule. Consistent with this prediction, Winter et al found that the abundance of human MID1 transcripts including exon 1c (an alternative exon that introduces a PTC) increased in the presence of the NMD inhibitor cycloheximide.38 Finally, Winter et al used RT-PCR to observe that different MID1 isoforms are produced in different tissues and at different developmental stages in both human and mouse. For instance, the PTC-introducing exon 1a was observed in five distinct transcripts in human fetal brain cells, two transcripts in fetal liver cells, and none in fetal fibroblasts. These results strongly suggest that alternative splicing and NMD are employed to regulate the overall abundance of productive MID1 transcripts.
Defects in regulating unproductive splicing can lead to disease. Myotonic dystrophy (DM), an autosomal dominant disease, is the most common form of adult-onset muscular dystrophy. DM has been shown to be caused by one of two repeat expansions, whose presence in mRNA sequesters several splicing factors39 including muscleblind, and thus induces splicing changes in several genes.40,41 Patients develop myotonia from lack of muscle-specific chloride channel 1 (ClC-1), which is misspliced in DM tissue.42 The normal developmental splice pattern for the ClC-1 transcript has a PTC in embryos but no PTC in adult cells. In DM tissue, ClC-1 transcript splicing reverts to its embryonic, PTC-containing splicing pattern. Consequently, ClC-1 mRNA is greatly reduced in abundance, likely due to the action of NMD.43 Thus, it appears that normal ClC-1 gene expression is governed by RUST, and that the DM disease is caused when this regulation is undermined by sequestration of splicing factors.
Autoregulatory Unproductive Splicing
There is abundant evidence that RUST is used for autoregulation. The autoregulated gene often, but not always, encodes a protein that is part of the splicing machinery. In some fascinating cases, proteins that are not generally involved in mRNA processing bind specifically to their own transcripts to affect splicing and elicit NMD. The clearest example of this is found not in a human gene but in yeast. Yeast genes are generally unspliced, but in the few intron-containing genes, intron inclusion can introduce an in-frame stop codon and target the transcript for NMD. The yeast ribosomal protein RPL30 binds to its own pre-mRNA to prevent the removal of a PTC-containing intron, which in turn triggers NMD.44 The mRNAs of other ribosomal protein genes, including RPL28 (CYH2) and RPS17B (RP51B), also sometimes retain their introns and become natural NMD targets, leaving open the possibility that their splicing is also regulated to elicit NMD.45
Some ribosomal proteins in Caenorhabditis elegans are similarly autoregulated. A screen for natural targets of NMD identified L3, L7a, L10a, and L12 ribosomal protein transcripts. Each of these transcripts can be alternatively spliced to generate either a productive isoform or an unproductive isoform that contains a PTC and is therefore degraded by NMD. The ratio of productive to unproductive alternative splicing of rpl-12 RNA is affected by levels of RPL-12 protein, indicating that unproductive splicing of rpl-12 RNA is under feedback control.46
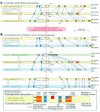
A striking number of splicing factors and elements of the splicing machinery are autoregulated through RUST (Fig. 5 and Table 1c,d). One such example is polypyrimidine tract binding protein (PTB), which inhibits splicing by competing with small nuclear (sn)RNA U2 associated factor (U2AF) for the polypyrimidine tract and perhaps through other mechanisms (reviewed in refs. 36 and 47). PTB RNA is alternatively spliced to produce two major productive isoforms (one of which lacks exon 9),48,49 one minor productive isoform lacking exons 3-9,49,50 and two unproductive isoforms lacking exon 11. Removing exon 11 causes a frameshift leading to a downstream PTC. PTB protein has been found to promote the removal of exon 11 from its own transcripts.49 Consequently, when PTB levels are high, PTB production is slowed by targeting PTB transcripts for NMD; and when PTB levels are low, production is accelerated by reducing the proportion of transcripts that are degraded.49,51,52
The CDC-like kinases (CLKs), which regulate an important family of splicing factors known as SR proteins, seem to be affected by RUST as well.16 RUST appears to regulate CLK1 levels through an indirect feedback mechanism. CLK1 has been shown to modify splicing of its own transcript indirectly, most likely through phosphorylation of SR proteins.53 Thus, as a variation of the autoregulatory circuit described above, increased CLK1 activity results in changes in the activity of one or more SR proteins. These SR proteins in turn affect the splicing of CLK1 pre-mRNA to favor a PTC+ transcript that is predicted to undergo NMD. This PTC+ transcript is stabilized by cycloheximide, consistent with its being normally degraded by NMD.54
RUST regulation of Clk1 levels may have a downstream effect on numerous SR proteins, and in turn on the splicing of many pre-mRNAs that are substrates of those SR proteins. Thus, alternative splicing can regulate factors that control splicing of other gene products.
Such a regulatory cascade of alternative splicing seems likely in the case of neuronal (n) PTB, a paralog of PTB that is regulated by RUST. There are reports that PTB affects the splicing of nPTB RNA, which is unsurprising given that PTB regulates the splicing of its own transcript, and nPTB is a close homolog of PTB. Furthermore, nPTB transcripts are alternatively spliced in a tissue-specific manner: in nonneuronal cells, PTC+ isoforms are preferentially produced but, in neurons, productive isoforms are made and translated. The resulting nPTB protein may compete with PTB for binding polypyrimidine tracts on heterologous pre-mRNAs, and may inhibit splicing more strongly or weakly than does PTB, resulting in an altered splicing pattern. Consequently, pre-mRNAs whose splicing pattern is sensitive to nPTB will be spliced differently in neurons than in other cell types.51
Transcripts encoding splicing factors that are autoregulated by RUST may also be subject to RUST that is triggered by heterologous factors; this is seen in the alternative splicing of PTB RNA, which can be affected by the splicing regulators raver1 and CELF4.49
Conservation of RUST
The coordinated use of alternative splicing and NMD is seen not only in mammals but in organisms as distant as yeast44 and possibly plants.55 The mechanism of PTC recognition differs in organisms other than mammals since it does not seem to depend on the location of the stop codon relative to exon junctions.56 (see chapters by Amrani and Jacobson, Behm-Ansmant and Izaurralde, and van Hoof and Green) There have been significant advances recently in elucidating the recognition mechanism in flies and yeast,57,58 but the rules are not clear enough to allow for computational identification of NMD targets. Nonetheless, NMD affects gene expression in a variety of different organisms.
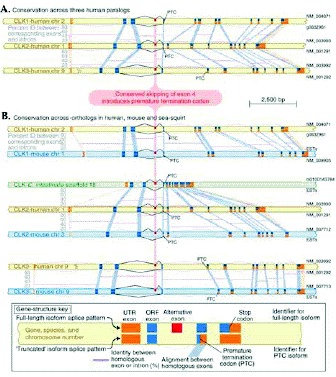
In several of the examples discussed above, analysis of orthologous and paralogous sequences suggests that splicing to generate PTC+ alternative isoforms, and thus RUST regulation, is conserved across species and across protein families. For PTB transcripts, the sequence and upstream regulatory elements of alternatively included PTC-containing exon 11 are very similar in transcripts that encode the Fugu rubripes ortholog as well as the human neuronal-specific paralog nPTB.49 Transcripts encoding mouse and monkey orthologs of the human multidrug resistance associated transporter ABCC4 share highly conserved PTC-containing exons that are orthologous to the alternatively included exons of the human ABCC4 transcript, another apparent RUST target.59 Particularly strong evidence of conservation of RUST is found in the Clk transcripts. Alternative splicing to exclude exon 4, introducing a frameshift and PTC, is conserved among transcripts encoding the three human paralogs (Clk1, Clk2, Clk3), the three mouse orthologs, and even the sole ortholog in the sea squirt Ciona intestinalis (Fig. 6).16
The action of NMD on some transcripts of a gene can be conserved even when the specific alternative splicing events that elicit NMD are not. As discussed above, MID1 RNA is a human RUST target. Interestingly, while PTC+ MID1 isoforms are found in human, mouse, and fugu, the responsible stop codons are introduced by alternative exons that show no homology between these species. Thus, in this case, it appears that the RUST mode of regulation was conserved while the specific sequence elements triggering it were not. This suggests that RUST is a generally useful mechanism that is easily applied to regulate expression of individual genes in organisms that already have both alternative splicing and NMD.
Why RUST?
A substantial portion of alternatively spliced mRNAs seem to be NMD targets. We have discussed possible explanations for the prevalence of unproductive splicing: do these splice forms represent biological noise, or are they produced to regulate protein expression? It is unlikely that all such splicing is used for regulation, but the growing body of examples presented above suggests that RUST plays a significant role in the cell.
Many truncated proteins encoded by alternative transcripts would be nonfunctional even if these transcripts were not removed by NMD. Is the combination of alternative splicing and NMD inherently different from alternative splicing that produces nonfunctional protein? Or does alternative splicing alone provide the important regulatory step, with NMD acting only as a convenient but unnecessary cleanup mechanism? Some proven cases of RUST illustrate that, in fact, the coordinated action of both pathways is required for regulation. The SR protein SC35 is autoregulated by RUST; its alternative splicing occurs in the 3' UTR to create an exon junction downstream of the original stop codon, without changing the open reading frame.60 The alternative splicing seems to have no role other than to cause the original termination codon to elicit NMD. Without NMD, the alternative mRNA would still encode the correct protein, so the alternative splice event alone could not be used to regulate protein levels. It seems, then, that some genes have evolved to take advantage of the combination of alternative splicing and NMD in a role different from that filled by either process alone.
RUST seems, at first, to be a wasteful process. A gene is transcribed and spliced, only to be degraded before it can produce a protein. Yet, we know that there are functional cases of RUST. The cost to the cell of transcribing apparently extraneous RNA is clearly not prohibitive. In humans, roughly 85-95% of transcribed sequence is spliced out as introns and discarded.61 Evidently, transcription of intronic sequences is not a significant selective disadvantage, and intron splicing may even provide some general selective advantage. Similarly, the cost of transcribing a pre-mRNA only to splice it into an unproductive form must be balanced by the advantages of an additional layer of regulation of gene expression.
How is a process like RUST beneficial to the cell? Transcriptional regulation is the most-studied means of controlling gene expression, but in some cases, additional control may be beneficial. Because splicing regulation occurs after the decision to transcribe a region, RUST may provide a rapid way to change the levels of productive mRNA. In extreme cases such as the dystrophin gene, the synthesis of a single transcript can take many hours,62 and the requirements of the cell might change after transcription begins but before a critical splicing decision that determines whether or not to introduce a PTC. Even when temporal regulation is not necessary, an extra layer of regulation can help fine-tune gene expression.
RUST is distinctive, in that it can either increase or decrease protein expression. The splicing factor PTB illustrates this point. At steady state, 20% of PTB pre-mRNA is spliced to an unproductive form.49 In general, we expect that a RUST-regulated gene is transcribed to produce more pre-mRNA than is needed at steady state, and that under normal conditions there is a base level of downregulation by unproductive splicing. This fraction of “wasted” transcripts constitutes the headroom available to the regulatory system to increase levels of productive transcript.
The prevalence of PTC+ alternative splice forms suggests a possible evolutionary interaction between alternative splicing and NMD.63 The existence of NMD could have led to an increase in alternative splicing. Any splicing errors that introduced PTCs would be removed by NMD, reducing the harmful effects of missplicing. As a result, the pressure to recognize splice sites perfectly would be lowered. Functional alternative splice forms could arise through splicing errors and then become established by sequence changes that strengthen their splice sites or add regulatory elements.
In a system with prevalent alternative splicing, regulation by RUST may evolve easily. For any particular gene, there are many possible alternative splicing events that could elicit NMD, including exon skipping, splicing within the 3' UTR, or recognition of cryptic splice sites. If the sequence of a gene changes slightly to promote one of these splicing events under certain splicing environments, and the resulting downregulation of gene expression by NMD is beneficial, then a basic sort of regulation has evolved. This has clearly occurred independently many times. Without NMD, alternative splicing can still regulate gene expression by producing nonfunctional proteins. The additional advantages of coupling splicing with NMD may be that it prevents accumulation of potentially harmful truncated proteins and that it reduces wasted translation, making unproductive splicing less costly.
Splicing factors such as PTB seem to be overrepresented among the known RUST targets. Is this a coincidence, or is RUST in fact used most often to regulate a small set of proteins that are already capable of binding pre-mRNAs? A protein that has an existing role in splicing may evolve autoregulation through splicing more easily than a protein that does not bind RNA. There are only a handful of known cases in which a protein that is not a splicing factor is autoregulated by RUST, and even these are predominantly ribosomal proteins that do bind RNA in other, nonsplicing contexts. However, autoregulation is by no means the only role of RUST, and there is no reason for nonautoregulatory RUST to affect splicing factors preferentially. The examples listed in Table 1 indicate that RUST is involved in the regulation of a diverse set of proteins.
The potential for alternative splicing to regulate gene expression has been appreciated for many years. Bingham et al proposed that “on/off regulation at the level of splicing might be unexpectedly common,” in a 1988 review featuring three cases of unproductive splicing in Drosophila melanogaster.64 An early paper about the splicing factor ASF discussed alternative splicing as a means to control gene expression.65 NMD adds an additional layer to the story;66 many of the unproductive splice forms identified years ago are now known to be degraded rather than translated. The prevalence of NMD-targeted splice forms has only recently become clear. Alternative splicing and NMD are often combined in an elegant way to regulate the expression of a wide range of genes. RUST seems to be a generally applicable, widespread, and readily evolved regulatory mechanism.
Acknowledgements
We would like to thank Richard E. Green and Rajiv Bhatnagar for helpful discussions and assistance with a few sections of the text. This work was supported by National Institutes of Health grants K22 HG00056 and R01 GM071655, and an IBM SUR grant. DAWS is supported by a predoctoral fellowship from the Howard Hughes Medical Institute. LFL is supported by NIH training grant T32 GM07127l. SEB is a Searle Scholar (I-L-110).
References
- 1.
- Boue S, Letunic I, Bork P. Alternative splicing and evolution. Bioessays. 2003;25:1031–4. [PubMed: 14579243]
- 2.
- Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30:13–9. [PubMed: 11753382]
- 3.
- Maniatis T, Tasic B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature. 2002;418:236–43. [PubMed: 12110900]
- 4.
- Garcia J, Gerber SH, Sugita S. et al. A conformational switch in the Piccolo C2A domain regulated by alternative splicing. Nat Struct Mol Biol. 2004;11:45–53. [PubMed: 14718922]
- 5.
- Resch A, Xing Y, Modrek B. et al. Assessing the impact of alternative splicing on domain interactions in the human proteome. J Proteome Res. 2004;3:76–83. [PubMed: 14998166]
- 6.
- Xing Y, Xu Q, Lee C. Widespread production of novel soluble protein isoforms by alternative splicing removal of transmembrane anchoring domains. FEBS Lett. 2003;555:572–8. [PubMed: 14675776]
- 7.
- Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem Sci. 1998. pp. 198–9. [PubMed: 9644970]
- 8.
- Lejeune F, Maquat LE. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr Opin Cell Biol. 2005;17:309–15. [PubMed: 15901502]
- 9.
- Cali BM, Anderson P. mRNA surveillance mitigates genetic dominance in Caenorhabditis elegans. Mol Gen Genet. 1998;260:176–84. [PubMed: 9862469]
- 10.
- Maquat LE, Carmichael GG. Quality control of mRNA function. Cell. 2001;104:173–6. [PubMed: 11207359]
- 11.
- Mendell JT, Sharifi NA, Meyers JL. et al. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–8. [PubMed: 15448691]
- 12.
- Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA. 2003;100:189–92. [PMC free article: PMC140922] [PubMed: 12502788]
- 13.
- Pruitt KD, Maglott DR. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 2001;29:137–40. [PMC free article: PMC29787] [PubMed: 11125071]
- 14.
- Boguski MS, Lowe TM, Tolstoshev CM. dbEST—database for “expressed sequence tags” Nat Genet. 1993;4:332–3. [PubMed: 8401577]
- 15.
- Boeckmann B, Bairoch A, Apweiler R. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–70. [PMC free article: PMC165542] [PubMed: 12520024]
- 16.
- Hillman RT, Green RE, Brenner SE. An unappreciated role for RNA surveillance. Genome Biol. 2004;5:R8. [PMC free article: PMC395752] [PubMed: 14759258]
- 17.
- Baek D, Green P. Nonsense-mediated decay, sequence conservation, and relative isoform frequencies in evolutionarily conserved alternative splicing. Proc Natl Acad Sci USA. 2005 [PMC free article: PMC1192826] [PubMed: 16123126]
- 18.
- Ishigaki Y, Li X, Serin G. et al. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell. 2001;106:607–17. [PubMed: 11551508]
- 19.
- Maquat LE. Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. [PubMed: 15040442]
- 20.
- Cao D, Parker R. Computational modeling and experimental analysis of nonsense-mediated decay in yeast. Cell. 2003;113:533–45. [PubMed: 12757713]
- 21.
- Chang AC, Sohlberg B, Trinkle-Mulcahy L. et al. Alternative splicing regulates the production of ARD-1 endoribonuclease and NIPP-1, an inhibitor of protein phosphatase-1, as isoforms encoded by the same gene. Gene. 1999;240:45–55. [PubMed: 10564811]
- 22.
- Veldhoen N, Metcalfe S, Milner J. A novel exon within the mdm2 gene modulates translation initiation in vitro and disrupts the p53-binding domain of mdm2 protein. Oncogene. 1999;18:7026–33. [PubMed: 10597303]
- 23.
- Zhang J, Maquat LE. Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J. 1997;16:826–33. [PMC free article: PMC1169683] [PubMed: 9049311]
- 24.
- Chester A, Somasekaram A, Tzimina M. et al. The apolipoprotein B mRNA editing complex performs a multifunctional cycle and suppresses nonsense-mediated decay. EMBO J. 2003;22:3971–82. [PMC free article: PMC169042] [PubMed: 12881431]
- 25.
- Inácio A, Silva AL, Pinto J. et al. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J Biol Chem. 2004;279:32170–80. [PubMed: 15161914]
- 26.
- Danckwardt S, Neu-Yilik G, Thermann R. et al. Abnormally spliced beta-globin mRNAs: A single point mutation generates transcripts sensitive and insensitive to nonsense-mediated mRNA decay. Blood. 2002;99:1811–6. [PubMed: 11861299]
- 27.
- Dreumont N, Maresca A, Boisclair-Lachance JF. et al. A minor alternative transcript of the fumarylacetoacetate hydrolase gene produces a protein despite being likely subjected to nonsense-mediated mRNA decay. BMC Mol Biol. 2005;6:1. [PMC free article: PMC546004] [PubMed: 15638932]
- 28.
- McAdams HH, Arkin A. It's a noisy business! Genetic regulation at the nanomolar scale. Trends Genet. 1999;15:65–9. [PubMed: 10098409]
- 29.
- Lareau LF, Green RE, Bhatnagar RS. et al. The evolving roles of alternative splicing. Curr Opin Struct Biol. 2004;14:273–82. [PubMed: 15193306]
- 30.
- Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat Genet. 2003;34:177–80. [PubMed: 12730695]
- 31.
- Kan Z, States D, Gish W. Selecting for functional alternative splices in ESTs. Genome Res. 2002;12:1837–45. [PMC free article: PMC187565] [PubMed: 12466287]
- 32.
- Letunic I, Copley RR, Bork P. Common exon duplication in animals and its role in alternative splicing. Hum Mol Genet. 2002;11:1561–7. [PubMed: 12045209]
- 33.
- Jones RB, Wang F, Luo Y. et al. The nonsense-mediated decay pathway and mutually exclusive expression of alternatively spliced FGFR2IIIb and -IIIc mRNAs. J Biol Chem. 2001;276:4158–67. [PubMed: 11042206]
- 34.
- Horikawa Y, Oda N, Cox NJ. et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet. 2000;26:163–75. [PubMed: 11017071]
- 35.
- Green RE, Lewis BP, Hillman RT. et al. Widespread predicted nonsense-mediated mRNA decay of alternatively-spliced transcripts of human normal and disease genes. Bioinformatics. 2003;19(Suppl 1):i118–21. [PubMed: 12855447]
- 36.
- Black DL. Mechanisms of alternative premessenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. [PubMed: 12626338]
- 37.
- Wagner EJ, Baraniak AP, Sessions OM. et al. Characterization of the intronic splicing silencers flanking FGFR2 exon IIIb. J Biol Chem. 2005 [PubMed: 15684416]
- 38.
- Winter J, Lehmann T, Krauss S. et al. Regulation of the MID1 protein function is fine-tuned by a complex pattern of alternative splicing. Hum Genet. 2004;114:541–52. [PubMed: 15057556]
- 39.
- Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–41. [PubMed: 9563950]
- 40.
- Brook JD, McCurrach ME, Harley HG. et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. [PubMed: 1310900]
- 41.
- Liquori CL, Ricker K, Moseley ML. et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–7. [PubMed: 11486088]
- 42.
- Charlet-B N, Savkur RS, Singh G. et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. [PubMed: 12150906]
- 43.
- Mankodi A, Takahashi MP, Jiang H. et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. [PubMed: 12150905]
- 44.
- Vilardell J, Chartrand P, Singer RH. et al. The odyssey of a regulated transcript. RNA. 2000;6:1773–80. [PMC free article: PMC1370047] [PubMed: 11142377]
- 45.
- He F, Peltz SW, Donahue JL. et al. Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1- mutant. Proc Natl Acad Sci USA. 1993;90:7034–8. [PMC free article: PMC47070] [PubMed: 8346213]
- 46.
- Mitrovich QM, Anderson P. Unproductively spliced ribosomal protein mRNAs are natural targets of mRNA surveillance in C. elegans. Genes Dev. 2000;14:2173–84. [PMC free article: PMC316897] [PubMed: 10970881]
- 47.
- Valcárcel J, Gebauer F. Post-transcriptional regulation: The dawn of PTB. Curr Biol. 1997;7:R705–8. [PubMed: 9382788]
- 48.
- Ghetti A, Piñol-Roma S, Michael WM. et al. hnRNP I, the polypyrimidine tract-binding protein: Distinct nuclear localization and association with hnRNAs. Nucleic Acids Res. 1992;20:3671–8. [PMC free article: PMC334017] [PubMed: 1641332]
- 49.
- Wollerton MC, Gooding C, Wagner EJ. et al. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell. 2004;13:91–100. [PubMed: 14731397]
- 50.
- Hamilton BJ, Genin A, Cron RQ. et al. Delineation of a novel pathway that regulates CD154 (CD40 ligand) expression. Mol Cell Biol. 2003;23:510–25. [PMC free article: PMC151525] [PubMed: 12509450]
- 51.
- Rahman L, Bliskovski V, Reinhold W. et al. Alternative splicing of brain-specific PTB defines a tissue-specific isoform pattern that predicts distinct functional roles. Genomics. 2002;80:245–9. [PubMed: 12213192]
- 52.
- Spellman R, Rideau A, Matlin A. et al. Regulation of alternative splicing by PTB and associated factors. Biochem Soc Trans. 2005;33:457–60. [PubMed: 15916540]
- 53.
- Duncan PI, Stojdl DF, Marius RM. et al. In vivo regulation of alternative pre-mRNA splicing by the Clk1 protein kinase. Mol Cell Biol. 1997;17:5996–6001. [PMC free article: PMC232448] [PubMed: 9315658]
- 54.
- Menegay HJ, Myers MP, Moeslein FM. et al. Biochemical characterization and localization of the dual specificity kinase CLK1. J Cell Sci. 2000;113(Pt 18):3241–53. [PubMed: 10954422]
- 55.
- Staiger D, Zecca L, Wieczorek Kirk DA. et al. The circadian clock regulated RNA-binding protein AtGRP7 autoregulates its expression by influencing alternative splicing of its own pre-mRNA. Plant J. 2003;33:361–71. [PubMed: 12535349]
- 56.
- Conti E, Izaurralde E. Nonsense-mediated mRNA decay: Molecular insights and mechanistic variations across species. Curr Opin Cell Biol. 2005;17:316–25. [PubMed: 15901503]
- 57.
- Amrani N, Ganesan R, Kervestin S. et al. A faux 3'-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–8. [PubMed: 15525991]
- 58.
- Gatfield D, Unterholzner L, Ciccarelli FD. et al. Nonsense-mediated mRNA decay in Drosophila: At the intersection of the yeast and mammalian pathways. EMBO J. 2003;22:3960–70. [PMC free article: PMC169044] [PubMed: 12881430]
- 59.
- Lamba JK, Adachi M, Sun D. et al. Nonsense mediated decay downregulates conserved alternatively spliced ABCC4 transcripts bearing nonsense codons. Hum Mol Genet. 2003;12:99–109. [PubMed: 12499391]
- 60.
- Sureau A, Gattoni R, Dooghe Y. et al. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 2001;20:1785–96. [PMC free article: PMC145484] [PubMed: 11285241]
- 61.
- Lander ES, Linton LM, Birren B. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. [PubMed: 11237011]
- 62.
- Tennyson CN, Klamut HJ, Worton RG. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet. 1995;9:184–90. [PubMed: 7719347]
- 63.
- Xing Y, Lee CJ. Negative selection pressure against premature protein truncation is reduced by alternative splicing and diploidy. Trends Genet. 2004;20:472–5. [PubMed: 15363899]
- 64.
- Bingham PM, Chou TB, Mims I. et al. On/off regulation of gene expression at the level of splicing. Trends Genet. 1988;4:134–8. [PubMed: 2853467]
- 65.
- Ge H, Zuo P, Manley JL. Primary structure of the human splicing factor ASF reveals similarities with Drosophila regulators. Cell. 1991;66:373–82. [PubMed: 1855257]
- 66.
- Hilleren P, Parker R. Mechanisms of mRNA surveillance in eukaryotes. Annu Rev Genet. 1999;33:229–60. [PubMed: 10690409]
- 67.
- Hovhannisyan RH, Carstens RP. A novel intronic cis element, ISE/ISS-3, regulates rat fibroblast growth factor receptor 2 splicing through activation of an upstream exon and repression of a downstream exon containing a noncanonical branch point sequence. Mol Cell Biol. 2005;25:250–63. [PMC free article: PMC538792] [PubMed: 15601847]
- 68.
- Carter MS, Li S, Wilkinson MF. A splicing-dependent regulatory mechanism that detects translation signals. EMBO J. 1996;15:5965–75. [PMC free article: PMC452383] [PubMed: 8918474]
- 69.
- Gudikote JP, Wilkinson MF. T-cell receptor sequences that elicit strong down-regulation of premature termination codon-bearing transcripts. EMBO J. 2002;21:125–34. [PMC free article: PMC125808] [PubMed: 11782432]
- 70.
- Wang J, Vock VM, Li S. et al. A quality control pathway that down-regulates aberrant T-cell receptor (TCR) transcripts by a mechanism requiring UPF2 and translation. J Biol Chem. 2002;277:18489–93. [PubMed: 11889124]
- 71.
- Skandalis A, Uribe E. A survey of splice variants of the human hypoxanthine phosphoribosyl transferase and DNA polymerase beta genes: Products of alternative or aberrant splicing? Nucleic Acids Res. 2004;32:6557–64. [PMC free article: PMC545452] [PubMed: 15601998]
- 72.
- Confaloni A, Crestini A, Albani D. et al. Rat nicastrin gene: cDNA isolation, mRNA variants and expression pattern analysis. Brain Res Mol Brain Res. 2005;136:12–22. [PubMed: 15893582]
- 73.
- Blanchette M, Labourier E, Green RE. et al. Genome-wide analysis reveals an unexpected function for the drosophila splicing factor U2AF(50) in the nuclear export of intronless mRNAs. Mol Cell. 2004;14:775–86. [PubMed: 15200955]
- 74.
- Cazalla D, Newton K, Cáceres JF. A novel SR-related protein is required for the second step of pre-mRNA splicing. Mol Cell Biol. 2005;25:2969–80. [PMC free article: PMC1069619] [PubMed: 15798186]
- 75.
- Henscheid KL, Shin DS, Cary SC. et al. The splicing factor U2AF65 is functionally conserved in the thermotolerant deep-sea worm Alvinella pompejana. Biochim Biophys Acta. 2005 [PubMed: 15777616]
- 76.
- Lallena MJ, Chalmers KJ, Llamazares S. et al. Splicing regulation at the second catalytic step by Sex-lethal involves 3'splice site recognition by SPF45. Cell. 2002;109:285–96. [PubMed: 12015979]
- 77.
- Pacheco TR, Gomes AQ, Barbosa-Morais NL. et al. Diversity of vertebrate splicing factor U2AF35: Identification of alternatively spliced U2AF1 mRNAS. J Biol Chem. 2004;279:27039–49. [PubMed: 15096518]
- 78.
- Engebrecht JA, Voelkel-Meiman K, Roeder GS. Meiosis-specific RNA splicing in yeast. Cell. 1991;66:1257–68. [PubMed: 1840507]
- 79.
- Petruzzella V, Panelli D, Torraco A. et al. Mutations in the NDUFS4 gene of mitochondrial complex I alter stability of the splice variants. FEBS Lett. 2005 [PubMed: 15975579]
- 80.
- Tsyba L, Skrypkina I, Rynditch A. et al. Alternative splicing of mammalian Intersectin 1: Domain associations and tissue specificities. Genomics. 2004;84:106–13. [PubMed: 15203208]
- 81.
- Screaton GR, Xu XN, Olsen AL. et al. LARD: A new lymphoid-specific death domain containing receptor regulated by alternative pre-mRNA splicing. Proc Natl Acad Sci USA. 1997;94:4615–9. [PMC free article: PMC20772] [PubMed: 9114039]
- 82.
- Colwill K, Pawson T, Andrews B. et al. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–75. [PMC free article: PMC449941] [PubMed: 8617202]
- 83.
- Duncan PI, Stojdl DF, Marius RM. et al. The Clk2 and Clk3 dual-specificity protein kinases regulate the intranuclear distribution of SR proteins and influence pre-mRNA splicing. Exp Cell Res. 1998;241:300–8. [PubMed: 9637771]
- 84.
- Le Guiner C, Gesnel MC, Breathnach R. TIA-1 or TIAR is required for DT40 cell viability. J Biol Chem. 2003;278:10465–76. [PubMed: 12533540]
- 85.
- Morrison M, Harris KS, Roth MB. Smg mutants affect the expression of alternatively spliced SR protein mRNAs in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1997;94:9782–5. [PMC free article: PMC23268] [PubMed: 9275202]
- 86.
- Wilson GM, Sun Y, Sellers J. et al. Regulation of AUF1 expression via conserved alternatively spliced elements in the 3' untranslated region. Mol Cell Biol. 1999;19:4056–64. [PMC free article: PMC104365] [PubMed: 10330146]
- 87.
- Wilson GM, Brewer G. The search for trans-acting factors controlling messenger RNA decay. Prog Nucleic Acid Res Mol Biol. 1999;62:257–91. [PubMed: 9932457]
- 88.
- Stoilov P, Daoud R, Nayler O. et al. Human tra2-beta1 autoregulates its protein concentration by influencing alternative splicing of its pre-mRNA. Hum Mol Genet. 2004;13:509–24. [PubMed: 14709600]
- 89.
- Condorelli DF, Nicoletti VG, Barresi V. et al. Structural features of the rat GFAP gene and identification of a novel alternative transcript. J Neurosci Res. 1999;56:219–28. [PubMed: 10336251]
- 90.
- Yoshikawa T, Makino S, Gao XM. et al. Splice variants of rat TR4 orphan receptor: Differential expression of novel sequences in the 5'-untranslated region and C-terminal domain. Endocrinology. 1996;137:1562–71. [PubMed: 8612486]
- 91.
- Moreau P, Carosella E, Teyssier M. et al. Soluble HLA-G molecule. An alternatively spliced HLA-G mRNA form candidate to encode it in peripheral blood mononuclear cells and human trophoblasts. Hum Immunol. 1995;43:231–6. [PubMed: 7558941]
- 92.
- Lui VC, Ng LJ, Sat EW. et al. Extensive alternative splicing within the amino-propeptide coding domain of alpha2(XI) procollagen mRNAs. Expression of transcripts encoding truncated pro-alpha chains. J Biol Chem. 1996;271:16945–51. [PubMed: 8663204]
- 93.
- Kolpakova E, Rusten TE, Olsnes S. Characterization and tissue expression of acidic fibroblast growth factor binding protein homologue in Drosophila melanogaster. Gene. 2003;310:185–91. [PubMed: 12801646]
- 94.
- Holder E, Maeda M, Bies RD. Expression and regulation of the dystrophin Purkinje promoter in human skeletal muscle, heart, and brain. Hum Genet. 1996;97:232–9. [PubMed: 8566960]
- 95.
- Jumaa H, Nielsen PJ. The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J. 1997;16:5077–85. [PMC free article: PMC1170142] [PubMed: 9305649]
- 96.
- Ram D, Ziv E, Lantner F. et al. Stage-specific alternative splicing of the heat-shock transcription factor during the life-cycle of Schistosoma mansoni. Parasitology. 2004;129:587–96. [PubMed: 15552403]
- 97.
- Wollerton MC, Gooding C, Robinson F. et al. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA. 2001;7:819–32. [PMC free article: PMC1370133] [PubMed: 11421360]
- 98.
- Besançon R, Prost AL, Konecny L. et al. Alternative exon 3 splicing of the human major protein zero gene in white blood cells and peripheral nerve tissue. FEBS Lett. 1999;457:339–42. [PubMed: 10471804]
- 99.
- Cellier M, Govoni G, Vidal S. et al. Human natural resistance-associated macrophage protein: cDNA cloning, chromosomal mapping, genomic organization, and tissue-specific expression. J Exp Med. 1994;180:1741–52. [PMC free article: PMC2191750] [PubMed: 7964458]
- 100.
- Dür S, Krause K, Pluntke N. et al. Gene structure and expression of the mouse APOBEC-1 complementation factor: Multiple transcriptional initiation sites and a spliced variant with a premature stop translation codon. Biochim Biophys Acta. 2004;1680:11–23. [PubMed: 15451168]
- 101.
- Niimi T, Copeland NG, Gilbert DJ. et al. Cloning, expression, and chromosomal localization of the mouse gene (Scgb3a1, alias Ugrp2) that encodes a member of the novel uteroglobin-related protein gene family. Cytogenet Genome Res. 2002;97:120–7. [PubMed: 12438750]
- 102.
- Dear TN, Kefford RF. The WDNM1 gene product is a novel member of the ‘four-disulphide core’ family of proteins. Biochem Biophys Res Commun. 1991;176:247–54. [PubMed: 2018519]
- 103.
- Golovkin M, Reddy AS. Structure and expression of a plant U1 snRNP 70K gene: Alternative splicing of U1 snRNP 70K pre-mRNAs produces two different transcripts. Plant Cell. 1996;8:1421–35. [PMC free article: PMC161266] [PubMed: 8776903]
- 104.
- Hornig H, Fischer U, Costas M. et al. Analysis of genomic clones of the murine U1RNA-associated 70-kDa protein reveals a high evolutionary conservation of the protein between human and mouse. Eur J Biochem. 1989;182:45–50. [PubMed: 2525092]
- 105.
- Spritz RA, Strunk K, Surowy CS. et al. Human U1-70K ribonucleoprotein antigen gene: Organization, nucleotide sequence, and mapping to locus 19q13.3. Genomics. 1990;8:371–9. [PubMed: 2147422]
- 106.
- Zhu S, Li W, Cao Z. A naturally occurring noncoding fusion transcript derived from scorpion venom gland: Implication for the regulation of scorpion toxin gene expression. FEBS Lett. 2001;508:241–4. [PubMed: 11718723]
- 107.
- Lemaire M, Prime J, Ducommun B. et al. Evolutionary conservation of a novel splice variant of the Cds1/CHK2 checkpoint kinase restricted to its regulatory domain. Cell Cycle. 2004;3:1267–70. [PubMed: 15467464]
- 108.
- Marshall B, Isidro G, Boavida MG. Naturally occurring splicing variants of the hMSH2 gene containing nonsense codons identify possible mRNA instability motifs within the gene coding region. Biochim Biophys Acta. 1996;1308:88–92. [PubMed: 8765755]
- 109.
- Barros SA, Tennant RW, Cannon RE. Molecular structure and characterization of a novel murine ABC transporter, Abca13. Gene. 2003;307:191–200. [PubMed: 12706902]
- 110.
- van Dam A, Winkel I, Zijlstra-Baalbergen J. et al. Cloned human snRNP proteins B and B' differ only in their carboxy-terminal part. EMBO J. 1989;8:3853–60. [PMC free article: PMC402073] [PubMed: 2531083]
Publication Details
Author Information and Affiliations
Authors
David A.W. Soergel, Liana F. Lareau, and Steven E Brenner*.Affiliations
Copyright
Publisher
Landes Bioscience, Austin (TX)
NLM Citation
Soergel DAW, Lareau LF, Brenner SE. Regulation of Gene Expression by Coupling of Alternative Splicing and NMD. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.