

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Trim-NHL proteins are defined by RING, B-Box and Coiled-coil protein motifs (referred to collectively as the Trim domain) coupled to an NHL domain. The C. elegans, D. melanogaster, mouse and human Trim-NHL proteins are potential and in several cases confirmed, E3 ubiquitin ligases. Current research is focused on identifying targets and pathways for Trim-NHL-mediated ubiquitination and in assessing the contribution of the NHL protein-protein interaction domain for function and specificity. Several Trim-NHL proteins were discovered in screens for developmental genes in model organisms; mutations in one of the family members, Trim32, cause developmental disturbances in humans. In most instances mutations that alter protein function map to the NHL domain. The NHL domain is a scaffold for the assembly of a translational repressor complex by the Brat proto-oncogene, a well-studied family member in Drosophila. The link to translational control is common to at least four Trim-NHLs that associate with miRNA pathway proteins. So far, two have been shown to repress (Mei-P26 and Lin41) and two to promote (NHL-2, Trim32) miRNA-mediated gene silencing. In this chapter we will describe structure-function relations for each of the proteins and then focus on the lessons being learned from these proteins about miRNA functions in development and in stem cell biology.
Introduction
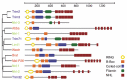
The year 2000 was an auspicious one for the miRNA field. In a series of three papers, the let-7 miRNA was first identified in a screen for mutations affecting developmental timing in C. elegans.1 Second, let-7 was quickly demonstrated to be highly conserved and expressed in a wide range of bilaterally symmetric animals,2 unlike the original miRNA gene, lin-4, that was thought to be restricted to C. elegans.3 Third, let-7 was shown to act as a repressor for a conserved gene, lin-41,1,4 within the developmental timing pathway. Analysis of predicted protein domains in LIN-41 led to the description of a novel class of so-called Ring, B-Box, Coiled-coil (RBCC) domain proteins.4,5 Later, the acronym Trim (Tripartite motif) was adopted for this sequence of protein domains. Current annotations recognize over 70 Trim domain proteins in mammalian genomes that can be subdivided into distinct classes based on the presence of additional C-terminal domains (reviewed in refs. 6,7). The C-terminus of LIN-41 was found to contain six copies of a 44 amino acid repeat sequence shared with two other known proteins: HT2A and NCL-1.4,5 These were the founding members of the Trim-NHL (NCL-1, HT2A, LIN-41) family. As seen in Figure 1, there are four Trim-NHL proteins encoded in the D. melanogaster, M. musculus and H. sapiens genomes and five in C. elegans. Several of these proteins, including the mouse ortholog of LIN-41, have recently been identified as regulators of miRNA pathway activity, the subject of this review.
There are several reviews of Trim domain proteins, including their structural characterization, disease phenotypes and functions in ubiquitin-mediated protein degradation.6-8 As of this writing, however, there is no comprehensive review of Trim-NHL family members and their roles in development and miRNA regulation. For this reason, we will begin with a brief outline of the common structural features of this protein family, followed by a review of earlier work on the functional characterization of each of the members. We will then turn to recent evidence that at least some of the Trim-NHL proteins act as post-transcriptional regulators of miRNA activity in their capacity as E3 ubiquitin ligases.
The Trim-NHL Family of Developmental Regulators
Although there have been a number of important studies on Trim-NHL proteins in chicken, zebrafish or rat, the bulk of the work has concerned the proteins from C. elegans, Drosophila, mouse and humans presented in Figure 1. The phylogenetic tree presented in Figure 1 is based on alignment of the C-terminal NHL sequences. Both the phylogenetic relationships and the individual domain structures of these proteins have been more rigorously treated elsewhere.6,7,9-11 As detailed below, the Trim-NHL sequence of motifs was first recognized in LIN-41 from C. elegans and consists of a RING-type (Really Interesting New Gene) Zinc-finger in close proximity to the N-terminus. One or two copies of a distinct Zinc-finger motif referred to as the B-Box follow the RING. The triad is completed by a hydrophobic heptad repeat termed the coiled-coil. This linear arrangement is quite constant, suggesting cooperative functional interactions between the three motifs. Less conserved is the Ig-filamin domain situated between the Trim motif and the NHL domain in several of the proteins. The paradigm NHL domain contains six copies of a 44 residue repetitive motif but the number of readily identified repeats varies from 2 to 6.
Each of these motifs will be discussed in turn, beginning with the RING domain. Although referred to as a RING finger, the actual structure is not fingerlike. Instead, the RING sequence contains four pairs of Zinc binding residues (numbered 1-4). The peptide chain loops back upon itself to juxtapose two non-adjacent pairs of Zinc binding residues (pairs 1 and 3) to form the first Zinc-coordination site. The peptide chain then reverses direction again to align the two remaining pairs (pairs 2 and 4). The result is a rigid, self-reinforced globular structure referred to as a "cross-brace" (reviewed in refs. 12,13; see NCBI Conserved Domain Database 00162). RING motifs are found in a large number of proteins and were originally thought to mediate DNA binding and/or protein-protein interactions. After several RING proteins were linked to ubiquitin-mediated protein degradation, the RING domain of the Cbl (Casitas B-lineage lymphoma) proto-oncogene was shown to actively participate in protein ubiquitination,14,15 a finding then extended to many additional family members16 (reviewed in refs. 13,17). The RING domain was later shown to possess intrinsic E3 ubiquitin ligase activity and to directly recruit and activate specific E2 ubiquitin conjugating enzymes.18,19 A brief description of the ubiquitin pathway will be provided in the next section (see Fig. 2). However, it seems certain that E3 activity is a general feature of the RING domain. Trim32 was the first Trim-NHL protein shown to be an E3 ubiquitin ligase,20,21 followed by Trim222 and mouse LIN-41 (referred to as either Trim71, mLin41, or Mlin41).23

Less is known about the B-Box motifs, which are found either singly or in tandem. When in tandem, the upstream B-Box conforms to a distinct consensus sequence. B-Boxes are most commonly associated with a RING domain, but as seen in Brat and Dappled/Wech this is not always the case (Fig. 1). Mutations in the B-Box for the nonNHL Trim proteins Trim5a and Trim18 (Midline-1) disrupt protein function (reviewed in ref. 6). As discussed below, mutations in the B-Box of human Trim32 lead to a distinct developmental disorder (Bardet-Biedl Syndrome)24 compared to mutations in the NHL domain (Limb Girdle Muscular Dystrophy).21,25 However, in these and in other cases the precise role of the B-Box remains elusive and may entail protein turnover, oligomerization or intracellular localization in addition to specific protein-protein interactions. The solution structure of the individual and the tandem B-Boxes of Trim18 is an important advance, revealing that both adopt a cross-brace conformation quite similar to the RING domain26-28 (see also NCBI Conserved Domain Database cl00034). This raises the interesting possibility that the B-Box may directly interact with the RING and participate in E3 activity, perhaps by serving as an accessory binding surface for E2 conjugating enzymes.
Of the three Trim motifs, the coiled-coil is the most widespread outside the Trim superfamily and represents a basic building block of protein structure (reviewed in refs. 18,29). In principle, coiled-coils can form higher order homo and heteromeric structures with a high degree of plasticity.29 Less is known about their structural or functional contribution to Trim domain function, although there is experimental evidence that they support homomeric interactions.9 The same study showed that multimerization mediated by the coiled-coil was required for correct localization to intracellular compartments. Trim2, Trim3 and Trim32 eGFP fusions were found to be cytoplasmic with a filamentous, diffuse or "speckled" distribution, respectively.9
When present, the Filamin homology domain, also referred to as an Ig-filamin repeat (see NCBI Conserved Domain Database cl02665), lies between the Trim motifs and the NHL repeats. No function has been assigned to the Filamin domain in the context of Trim-NHL family members. In Filamin proteins, the Ig-Filamin repeats are present in up to twenty-four copies and adopt a rod-like β-barrel conformation that interacts with actin and many other partners (see ref. 30 for a recent review).
Of course, the defining domain of the Trim-NHL family is the NHL repeat (Fig. 1). The similarity of the NHL repeats to a previously characterized motif, the WD-40 β-propeller, was significant enough to suggest a similar secondary structure.5 Furthermore, many of the genetically isolated mutations in LIN-41 and Brat mapped to the NHL domain.4,31 The NHL domain was necessary and sufficient for rescue of brat embryos during embryonic patterning,32 demonstrating that the isolated domain has activity as a translational repressor (see below for details). The crystal structure of the isolated NHL from Brat has been solved and revealed that each of the NHL repeats forms one "blade" of a six-bladed propeller, with the exception of the first blade. β-sheet elements derived from the first and sixth NHL repeat contribute to the first blade. This brings both ends of the structure together in a circular, or doughnut-like arrangement.33 Each blade is composed of four β-sheets connected by exposed loops. Structural modeling suggests that the RNA-binding protein Pumilio interacts with one face of the propeller via the looped out residues. The properties of this potential interaction surface are predicted to vary substantially among the individual Trim-NHL family members such as Dappled/Wech.33
The Trim Domain as E3 Ubiquitin Ligase
As noted above, E3 ubiquitin ligase activity has been documented for an increasing number of Trim proteins, making it the potentially largest class of E3 enzymes. In the case of Trim-NHL proteins, a role in protein ubiquitination could account for the diversity of their known functions in processes ranging from developmental timing (LIN-41, NHL-2), patterning (Brat), cell growth, division and proliferation (Brat, Mei-P26, NCL-1), endosomal trafficking (Trim3), muscle integrity (Trim32, Wech) and germ line (LIN-41, Mei-P26) or nervous system development and function (Brat, Mei-P26, mLin41, Trim2, Trim32). Ubiquitination is a post-translational modification rivaling phosphorylation in extent and consequence. The ubiquitin literature is far too vast to summarize here and there are many excellent reviews.13,34,35 Basic features relevant to this review are summarized in Figure 2. Covalent attachment of ubiquitin to substrate proteins involves a tag team of three enzymatic activities (E1-E3). First, in an ATP-consuming reaction ubiquitin is activated by attachment to an E1 activating enzyme. Interaction with an E2 conjugating enzyme leads to transfer of ubiquitin from the E1 to the active site of the E2. E3 ligases belong to one of two large classes, HECT or RING. In the case of RING domain E3s, the E3 serves as a bridge between the E2 and the substrate protein with primary responsibility for substrate specificity. Trim E3s are believed to stimulate catalysis by inducing proximity between the E2 and the substrate.13 A total of six known E1s (only two utilize ubiquitin, the others employ ubiquitin-like proteins such as SUMO—Small Ubiquitin-like MOdifier) can chose among ~40 E2s. A given E3 will productively interact with only one or a few E2s and can recognize multiple substrate proteins. Substrate ubiquitination can take many forms, including monoubiquitination, oligoubiquitination and various types of polyubiquitin chains (Fig. 2). To date, for the Trim-NHLs there is experimental evidence for polyubiquitination activity using Lys48 linkages, one of seven lysines available in ubiquitin for chain elongation.21-23,36,37 This is the classical signal for recognition and degradation by the 26S proteasome. Alternative linkages are nonproteolytic and alter substrate function, for example in directing protein traffic to vesicles (monoubiquitination) or by acting as a scaffold for protein-protein interactions (Lys63 polyubiquitination). Since linkage specificity is primarily a function of the E2, an E3 can have dual capacity and there is experimental support for substrate monoubiquitination via Trim32.38
Functional Analysis of Individual Trim-NHL Family Members
Trim32
Trim32 was the first Trim-NHL gene to be cloned, in a two-hybrid screen for proteins interacting with the HIV Tat protein.39 A missense mutation in the Trim32 NHL domain was later shown to underlie the human developmental disturbance Limb-girdle muscular dystrophy (LGMD2H, Type 2H).40 Subsequent studies have identified additional Trim32 mutations in LGMD2H; all map to the NHL and affect both the ability of the protein to self-interact and to bind E2.25 In keeping with the muscle phenotype, Trim32 was originally shown to physically interact with the head and neck region of the myosin heavy chain. The coiled-coil domain was required for myosin binding. Trim32 was shown to possess E3 activity in an in vitro autoubiquitination assay, in conjunction with the E2 enzymes UbcH5a, UbcH5c or UbcH6. Based on in vitro and in vivo assays, a role for Trim32 in actin degradation was proposed during muscle regeneration.21 More recently, Dysbindin was identified as an additional candidate for Trim32-mediated ubiquitination, primarily in the form of monoubiquitination.38 Dysbindin is a component of Bloc-1, an actin-associated protein complex involved in the biogenesis of lysosome-related vesicles.41
An alternative model was proposed for Trim32 in keratinocytes, in which the NHL domain of Trim32 interacts with Piasy. Piasy is itself an E3 ligase, but is specific for SUMO,36 an alternative ubiquitin-like protein tag with a distinct spectrum of activities. Interestingly, Trim32 not only targeted Piasy for ubiquitin-mediated degradation, it also caused the relocalization of Piasy from the nucleus to cytoplasmic foci resembling P-bodies. A consequence of Trim32 expression in keratinocytes was reduced sensitivity to NF- κB dependent apoptosis, suggesting a role for the Trim32/Piasy regulatory interaction in skin carcinogenesis.36
It is not surprising that an E3 ligase might have multiple substrates, with the possibility of multiple gene-phenotype relationships. This point is underscored by an independent mutation in the B-Box domain of Trim32 that predisposes for the Bardet-Biedl Syndrome.24 Bardet-Biedl is a multifaceted disorder encompassing renal abnormalities, retinal dystrophy, polydactyly, mental retardation and obesity. Nevertheless, there is no obvious phenotypic overlap between LGMD2H and Bardet-Biedl and none of the many Bardet-Biedl genes other than Trim32 have any known connection to muscular dystrophy. However, the finding that Bardet-Biedl genes as a group cause defects in the basal body of ciliated cells and impair ciliary function42,43 may be relevant to the proposed role for Trim32 in neurogenesis,37 which is discussed below. New insights are likely to be gained by transgenic approaches, for example deletion of Trim32 in mice was recently shown to phenocopy many of the myopathies associated with LGMD2H.44 The phenotype also has a neurogenic component, manifested biochemically by reduced levels of neurofilaments and morphologically by reduced diameters of motor axons. Bardet-Biedl symptoms were not described. One caveat for comparing the human mutations with the gene deletion model is that the mutations in Bardet-Biedl or in LGMD2H are not necessarily null alleles. Therefore, it may be necessary to generate mice carrying NHL or B-Box mutations to unravel the full functions of Trim32.
LIN-41
In C. elegans, lin-41 was discovered as a major downstream target for the let-7 miRNA in the developmental timing pathway1,4 (reviewed in ref. 45). Gain and loss of function lin-41 mutants displayed opposite phenotypes affecting vulval growth and morphology, oocyte production and the timing of cell cycle exit and terminal differentiation of a set of blast cells in the hypodermis known as seam cells.4 Genetic analysis gave the first indication that LIN-41 is itself a post-transcriptional regulator, acting to repress a transcription factor downstream of lin-41 in the timing pathway. Sequencing of lin-41 led to the recognition of the Trim-NHL domain structure as a new protein family, together with the previously cloned NCL-1 and HT2A/Trim32.4,5 Many of the null mutations recovered in the genetic screen mapped to the NHL domain, a clear indication of its functional importance. Ultimately, the presence of the Trim domain led to the annotation of mammalian homologs of LIN-41 as Trim71, but here we retain the designation Lin41 for the mouse and human proteins in deference to this pioneering work in C. elegans.
Database searches revealed the presence of LIN-41 homologs in Drosophila and in vertebrates.4 In zebrafish Kloosterman et al verified the prediction by Slack and coworkers that regulation of lin-41 by let-7 is conserved.46 Three papers described the cloning of the chicken, mouse and human genes and analyzed embryonic mRNA expression patterns.47-49 In both mouse and chicken, Lin41 was temporally regulated and inversely correlated with the induction of let-7 and miR-125 expression in embryonic development.48,49 In C. elegans, lin-41 is expressed in neurons, muscles and gonads.4 Muscle expression was confirmed in the chicken47 and all three research groups noted temporally dynamic regulation of the mRNA in the limb, wing and tail buds.47-49 Consistent with this, mLin41 expression was modified in mouse mutants for the limb bud patterning genes Sonic hedgehog, Fgf-8 and Gli3.47 This represents the first evidence for developmental control of Lin41 and its miRNA regulators let-7 and miR-125 by mammalian signaling pathways.
In Drosophila, the closest lin-41 homolog has been referred to as either dappled or, more recently, wech. As seen in Figure 1, the protein lacks a RING domain. Dappled refers to the appearance of mutant larvae, due to the presence of melanotic tumors that are thought to be a sign of tissue abnormalities.50 dappled was next encountered in a differential screen for genes expressed in the embryonic head. At Stage 11, the mRNA was expressed at low levels throughout the embryo and more strongly in neuroblasts, ganglion mother cells and neurons.51 A more comprehensive in situ study documented expression in the central nervous system (CNS) and peripheral nervous system (PNS) throughout embryogenesis.11 Ectopic dappled expression interfered with proper development of the eye and sensory organs.11,52 Although the significance of these findings for normal development remains unclear, the eye phenotype was sensitive to let-7 expression levels.11
A quite different view emerged from studies of integrins and muscle attachment53 (reviewed in refs. 54,55). Analysis of a mutant with a near complete loss of embryonic muscle attachment identified a loss of function mutation in a gene identical to dappled that the authors named wech. Loss of Wech had no obvious effect on muscle cell differentiation, as myoblasts fused and expressed actin and myosin heavy chain. The attachment defect could be rescued by specifically restoring Wech expression in muscle and tendon cells, or in either cell-type individually. The Wech protein colocalized to cortical foci in muscle cells with the adhesion proteins Talin, ILK (Integrin Linked Kinase) and Tensin. These three proteins act to link integrin to the actin cytoskeleton at focal adhesion points between cells known as hemiadherens junctions. Wech may serve as an adaptor protein in the organization of these foci, as ILK and Tensin localization was dependent on Wech and Wech in turn was dependent on integrin and Talin. Interestingly, the B-Box and coiled-coil domains were required for the interaction with both ILK and Tensin.
The adhesion junctions studied by Löer et al are by no means restricted to the muscle-tendon interface, but there is not yet any information on Wech function in other tissues. It may be relevant that Ambros and colleagues found that let-7 is critical for the attainment of an adult neuromusculature.56 let-7 was strongly upregulated in the Drosophila CNS and PNS during metamorphosis. High-level expression was also observed in adult body wall muscle and in remnants of larval muscle that are initially retained after metamorphosis. Consistent with this expression pattern, let-7 null mutants were viable without gross defects in external morphology but suffered from severe disturbances in motor function. Overall CNS development appeared to be normal, but a juvenile pattern of muscular innervation persisted that coincided with a failure of the muscular remodeling that normally occurs upon execution of metamorphosis.56 Determining the contribution of Wech/Dappled to this transformation is an obvious next step and it will be interesting to see if the protein is downregulated in neurons and muscle at the transition. If so, the question would then be whether another Trim-NHL protein adopts the larval function of Wech/Dappled in adults and what the functional consequences of such a switch might be.
Trim2 and Trim3
As seen in Figure 1, the mammalian Trim2 and Trim3 proteins are more closely related to one another than to their D. melanogaster or C. elegans paralogs. Like Trim32, both are strongly, but by no means exclusively, expressed in the brain. Originally referred to as Berp (Brain enriched Ring Protein) Trim3 was cloned in a search for RING finger proteins in the brain.57 Like Trim32, Trim3 was found to interact with myosin. Unlike Trim32, the NHL domain directed binding to the tail region of the unconventional Class V myosins Va and Vb. Class V myosins are primarily involved in transport of macromolecular complexes, organelles and vesicles, including endosomes (reviewed in ref. 58). Consistent with a role in endosomal trafficking, Trim3 was found to copurify with early endosomes and to form a complex with Actinin-4, Hrs and Myosin Vb dubbed CART (Cytoskeleton-Associated Recycling or Transport).59 Independently, Trim3 was found to copurify with the Lst2 protein.60 Focusing on Lst2, Mosesson et al found that Lst2 promotes trafficking of the receptor for epidermal growth factor (EGFR) to early endosomes. Furthermore, the localization of Lst2 to early endosomes was inhibited by monoubiquitination, but it is not clear if Lst2 is a substrate for Trim3.60 Mono- and oligoubiquitination are recurring signals in the complex pathways that decide receptor fate after endocytosis: either recycling by return to the cell membrane or degradation by sequential sorting to endosomes and then to lysosomes (reviewed in ref. 61). It remains to be seen if Trim3 has E3 activity, if it targets Lst2 and what its putative role in vesicle trafficking may be.
Trim2 was originally designated NARF (Neural Activity-related Ring Finger), because Trim2 mRNA was upregulated after pharmacological stimulation of seizure activity in rats. Like Trim3, the NHL domain of Trim2 interacted with Myosin V.62 Within the brain, Trim2 expression is highest in the CA1-3 neurons of the hippocampus,63 a finding confirmed using a gene-trap allele to record Trim2 (Trim2GT) promoter activity.22 Trim2 is dispensable for embryonic development, as mice homozygous for Trim2GT appeared normal until 1.5 months. The mice then presented a neurodegenerative phenotype initially manifested by tremor, followed by ataxia and seizures. Morphologically, Trim2GT brains displayed increased accumulation of neurofilament and abnormally swollen axons. This axonopathy preceded both neurological symptoms and loss of neurons in the cerebellum and retina. Biochemically, Trim2 was shown to interact with the E2 enzyme UbcH5a in an autoubiquitination assay,22 one of the E2 utilized by Trim32.21 However, unlike Trim32 no autoubiquitination activity was observed with Ubc5Hc or UbcH6. The light chain of neurofilament may be one substrate for Trim222 and it will be interesting to compare axonal phenotypes and ubiquitin substrates for Trim2 and Trim32.
Brat
The Drosophila protein Brat (Brain tumor) is perhaps the best studied of the Trim-NHL proteins, due to its dual role as an embryonic patterning gene and as a tumor suppressor in the larval brain. brat mutants also have defects in male and female fertility, cuticle formation and occasionally form melanotic tumors,64 a constellation reminiscent of lin-41 and dappled, respectively. During early embryonic development Brat is required for the establishment of an anterior-posterior gradient in the translation of hunchback (hb) mRNA, a maternal effect gene and Zinc finger transcription factor.32 In this role as translational regulator, Brat participates in complex formation with the translational regulator Pumilio and the sequence specific RNA-binding protein Nanos. Despite detailed information regarding the structural features of the regulatory complex, less is known about the mechanistic contribution of Brat to translational control.33 Sonoda and Wharton have suggested that Brat may assemble on multiple mRNAs by engaging in combinatorial interactions with RNA-binding proteins other than Nanos,32 but this has not yet been confirmed experimentally. This suggestion may, however, prove to be correct in light of the discovery that Brat interacts with the miRISC effector protein Ago1,65 as discussed in more detail in the section on Mei-P26.
The most striking feature of brat mutants is the growth of larval tumors in the optic centers that can attain ten times the normal size.64 Frank et al found that ectopic Brat inhibits RNA synthesis and cell proliferation while increasing cell size in several organ contexts.66 However, Brat is primarily expressed in the embryonic PNS, the ventral nerve cord and the developing brain.31 Even within the CNS, the tumor origin was later shown to be confined to a subset of neural progenitors67 with the characteristics of transit amplifying cells.68 A recent review of the relevant issues in the larval nervous system is highly recommended69 (see also ref. 70) and can only be briefly treated here. In the basic scheme, the intrinsic apical-basal polarity of neuroblasts is used as a guide to asymmetrically sort cell fate determinants to daughter cells (Fig. 3a). Different regions of the fly brain execute specific programs of proliferation and specification, but the basic scenario of asymmetric division is that the apical daughter retains neuroblast character while the basal daughter, called a ganglion mother cell (GMC), is smaller and destined for cell cycle exit and terminal differentiation. Segregation of cell fate determinants requires the action of apical proteins in the mother cell, including Par-3, Par-6 and atypical Protein Kinase C (reviewed in refs. 69,70)(Fig 3). These proteins are required for basal accumulation of a second set of proteins, which drive GMC specification. Brat is a recent entry into this group of specification factors, which includes Numb, an inhibitor of the Notch pathway and Prospero, a transcription factor that suppresses self-renewal and promotes neural differentiation.67,71,72 Brat was shown to bind Miranda,71,72 a scaffold protein that accumulates in a crescent at the basal pole of the mother cell destined for inheritance by the GMC (or basal neuroblast, see below). After division, Brat suppresses neuroblast markers and promotes cell cycle exit in the GMC.67,71,72 One proposed regulatory target for Brat is the Drosophila Myc protein, a neuroblast marker that is dysregulated in brat mutants.71 A new wrinkle in this basic model comes from the discovery that the tumor lineage in brat mutants does not immediately produce a GMC but instead a secondary neuroblast68 (Fig. 3b). This is a transit-amplifying cell that produces GMCs after further division. In this scenario, loss of Brat leads to tumors because the secondary neuroblast fails to mature. How Brat controls proliferation and maturation of these cells remains unclear, but it appears to work cooperatively and in parallel to Numb.68 Recent evidence derived from studies of Mei-P2665 and other Trim-NHLs to be discussed next points to a role for Brat as regulator of the miRNA pathway, but this has yet to be demonstrated.

Trim-NHL Proteins as Regulators of the miRNA Pathway
Mei-P26
Several of the biological processes relevant to the Drosophila mei-P26 gene have already been mentioned in relation to other Trim-NHL proteins: seizure (Trim2), gonad development (LIN-41) and centrosome function (Trim32). The mei-P26 gene was first encountered as recombination defective in a screen for mutants in meiosis.73 Further analysis extended the phenotype of mei-P26 mutants to both male and female sterility.74 Affected ovaries of the mutants contained inappropriately high numbers of poorly differentiated cells. Testes were cystic and failed to produce motile spermatozoa.74 Despite strong specificity for gonads in its zygotic expression, RNAi against mei-P26 resulted in disruption of embryonic CNS and PNS organization.75 Supporting a role in the CNS, mei-P26 was picked up in a mutational screen as a strong suppressor of seizures in an eas (easily shocked) background.76 eas encodes an ethanolamine kinase; the exact nature of seizure susceptibility is not known but the eas mutation affects phospholipid metabolism and neuroblast proliferation.77,78 Two missense mutations were discovered in the mei-P26 suppressor allele, one in the coiled-coil domain and one in the NHL domain.76
Little information on the molecular function of Mei-P26 was available until 2008, when the Knoblich laboratory first implicated Trim-NHL proteins in the regulation of the miRNA pathway.65 In visually stunning work, they first carefully dissected the growth control failure in the ovaries of mei-P26 mutants. Terminal differentiation to oocytes was blocked in mei-P26 ovaries, with an accumulation of cells in an intermediate state of differentiation in which proliferation markers (CyclinE, Phospho-histone-H3) were upregulated (see Fig. 3c). In analogy to their earlier work on Brat, Myc expression and nucleolar size were increased in the germ cell progeny of mei-P26 mutants. Conversely, overexpression of Mei-P26 led to depletion of ovarian germ cells. Mei-P26 expression was upregulated in the initial progeny of ovarian stem cells and Mei-P26 levels increased during the four rounds of transit amplifying proliferative cell divisions. High Mei-P26 levels direct cell cycle exit and entry into terminal differentiation (Fig. 3c).
The model for Mei-P26 function in the germ line developed by Neumüller et al shares similarity to the proposed role of LIN-41 in the vulva and seam cells4 and of Brat in neuroblasts.67,68,71,72 Support for a common mechanism came from the observation that Mei-P26 and Brat interact with Ago1,65 the Drosophila Argonaute family member responsible for miRNA-mediated gene silencing.79 The physical link between Mei-P26 and Ago1 was reflected in global miRNA expression levels: increased in mei-P26 mutants and decreased upon Mei-P26 overexpression.65 On balance, miRNAs appear to support ovarian stem cell self-renewal, based on the phenotypes of mutations that block miRNA biogenesis in the ovary.80,81 miRNA function was also affected, as Mei-P26 interfered with repression of target genes by the bantam miRNA. Suppression of bantam by Mei-P26 may be a significant part of the pathway, as bantam is required for germ cell self-renewal and mei-P26 is itself a predicted target for bantam. These results were the first indication that regulatory interactions between miRNAs and Trim-NHL proteins are a two-way street, which will be the theme of the rest of the review.
NHL-2
In a recent paper, the group of Victor Ambros set out to test if LIN-41 and other Trim-NHL proteins share functions in the C. elegans developmental timing pathway.82 Mutations in nhl-2 led to premature stem cell maturation in a manner reminiscent of lin-4 and let-7 family mutants (let-7, mir-48, mir-84, mir-241). Combining mutations in let-7 family members with the nhl-2 mutant exacerbated the phenotype, as would be expected if NHL-2 and let-7 cooperate. Furthermore, cooperativity was not limited to let-7 but was also seen for the unrelated miRNA lsy-6. Interestingly, loss of nhl-2 was found to partially rescue a lin-41 allele, suggesting that NHL-2 and LIN-41 might be actors in a single pathway, but with opposite roles. The demonstration that NHL-2 interacts with the CGH-1 protein in a two-hybrid screen strengthened the link to the miRNA pathway.82 CGH-1 is homologous with the human RCK/p54 protein, a Dead Box helicase that associates with Argonautes and the miRISC in P-bodies.83 NHL-2 was then shown to colocalize to P-bodies and to physically and genetically interact with the core miRISC proteins ALG-1 and AIN-1 (C. elegans Argonaute and GW182 orthologs, respectively). Despite the ability to enhance silencing of several miRNA target genes, NHL-2 did not increase miRNA expression levels, suggesting that NHL-2 enhances the efficiency of the miRISC downstream of miRNA biogenesis.82
Following the demonstration that Mei-P26 is an inhibitor of the miRNA pathway,37 the work by Hammell et al showed that NHL-2 has the opposite role as a positive regulator (reviewed in ref. 84). Mechanistically, the action of the two proteins has not yet been clarified, but several models for NHL-2 were discussed.82 Relying on the precedent for Brat as scaffold protein and the finding that the interaction between NHL-2 and the miRISC is RNase sensitive, one possibility is that NHL-2 might facilitate binding of CGH-1 to the miRISC effector-mRNA complex. This is consistent with the role of yeast and human orthologs of CGH-1 in promoting mRNA decapping and translational repression.85,86 Alternatively, action of NHL-2 as E3 ubiquitin ligase might serve to modify protein-protein interactions or activities within the miRISC. One interesting suggestion is that NHL-2 might fulfill a fail-safe function by ubiquitinating and promoting the degradation of any translation products that escape translational silencing. Insights into the mechanism of NHL-2 function should soon emerge, perhaps through studies that combine the power of C. elegans and D. melanogaster genetics with in vitro assays for miRISC activity.
Trim32 and the miRNA Pathway
Published back-to-back with the NHL-2 paper, the Knoblich lab reported that mouse Trim32 also functions as an activator of the miRNA pathway.37 Pursuing functional homologs of Brat in mammalian neurogenesis, Trim32 was first shown to suppress proliferation of heterologous cells. During cortical development, expression of Trim32 tracked cell cycle exit and neuronal differentiation both temporally and spatially. Like Brat, Trim32 was shown to distribute asymmetrically to the daughter cells after some, but not all, neural progenitor cell divisions. Asymmetric inheritance was most frequent at the high point of neurogenesis around E14.5 (see Fig. 3d). Consistent with this, cultured neural progenitors displayed symmetric Trim32 inheritance during proliferative divisions but switched to an asymmetric mode under conditions favoring neurogenesis. These findings suggest that neurogenic signals regulate Trim32 localization, although the nature of the signal or the mechanism causing asymmetric Trim32 accumulation has not been determined. Current discussions of the determinants of asymmetry and the relationship to cell fate in the mammalian neuroepithelium are complex and controversial and are beyond the scope of this review (refer to refs. 69,87,88). One clue was that Trim32 concentrates in the retracting basal fiber (also referred to as radial fiber or basal process), a structural feature of mammalian neural progenitors that is involved in the initiation of the cleavage furrow and perhaps asymmetric daughter cell fate (Fig. 3d) (reviewed in ref. 89).
To better characterize the role of Trim32 in neural progenitors, Schwamborn et al first combined in utero electroporation to manipulate Trim32 expression levels in the neuroepithelium in vivo with subsequent in vitro culture of the transfected cells to follow their fate.37 Trim32 overexpression reduced the proliferative capacity and enhanced neuronal marker expression of the transfected cells. Conversely, Trim32 knockdown led to enhanced proliferation and delay in in vitro neurogenesis. Similar results were obtained when cell fate was examined in vivo: after electroporation Trim32 overexpressing cells showed an increased rate of migration into the cortical layers; Trim32 knockdown cells were delayed in their exit from the progenitor zone. These results support a model in which Trim32 plays a similar role to Brat: asymmetric Trim32 inheritance might support cell cycle exit and favor neuronal fate choice.
To extend the parallel to Brat, Schwamborn et al next demonstrated that Trim32 inhibits c-Myc. c-Myc protein levels were reduced in cells overexpressing Trim32, accompanied by accumulation of polyubiquitinated c-Myc. Both effects were eliminated after mutation of the RING domain in Trim32, suggesting that Trim32 might directly target c-Myc for degradation. A second parallel to Brat (and Mei-P26) was then explored, with the demonstration that Trim32 interacts with Ago1 in immunoprecipitation assays. Comparison of the signature of miRNAs that coprecipitate with Trim32 or Ago1 identified eight miRNAs with significant enrichment in Trim32 complexes. One of the significant hits was let-7a (but not other let-7 family members). To test a role for let-7a in neuronal differentiation, let-7a activity was blocked by in utero delivery of an anti-let-7a LNA antagomir. Cells receiving the let-7a antagomir had an approximately two-fold higher likelihood of retaining the progenitor marker Nestin. Conversely, overexpression of let-7a in the progenitor zone led to a strong increase in neuronal differentiation, as indicated by neuronal marker expression. This result suggests that Trim32 acts in part by increasing the activity of let-7a in neural progenitors.37
Like many landmark papers, the work of Schwamborn and coworkers raises as many new questions as it answers. Given the central role that c-Myc and let-7 play in the circuitry of stem cells, with Myc acting as a suppressor of let-7 transcription while also being a direct target gene for let-7 (reviewed in ref. 90, see also ref. 91), it is unclear if the regulatory interactions with Myc and let-7 represent independent activities of Trim32. It would be interesting to confirm if c-Myc and Trim32 directly associate and in what cellular compartment the interaction takes place. Does Trim32 draw Myc into P-bodies, as has been suggested for Piasy?36 It would also be interesting to know if the effect of Trim32 on c-Myc degradation extends to other Myc family members, including N-Myc, a protein more directly tied to neurogenesis than c-Myc.92,93 Also, if downregulation of Myc is central to both Brat and Trim32, is the RING-less Brat able to indirectly mediate d-Myc degradation? This question can be stated more broadly, since it is unclear if the E3 activity of Trim32, as well as NHL-2, is required for enhancing miRNA pathway activity. Schwamborn et al reported that the RING domain of Trim32 was not required to suppress let-7 activity in a reporter assay, but it is far from clear if c-Myc is the only relevant target for Trim32 in suppressing cell proliferation and encouraging neuronal differentiation in vivo.37 Another intriguing possibility is whether or not asymmetric Brat and Trim32 segregation to daughter cells implies differential inheritance of miRNAs and the core miRISC machinery. Does Trim32 pull neurogenic miRNAs and an activated miRISC into the cell destined to adopt a neuronal fate? It would be very interesting to compare the activity of reporters for Trim32-associated miRNAs before and after symmetrical and asymmetrical cell divisions. Such a mechanism might have functional relevance, as several studies reported that neural progenitors express let-7 prior to terminal differentiation.94-96 Finally, it is not yet known if the Trim32-Ago1 interaction is unique or common to all Argonautes. This would be important in assessing the significance of preferential sorting of miRNAs to miRNPs containing Trim32. It is not currently known how functionally redundant mammalian Argonautes are and if they favor some miRNAs over others. Therefore, loading of miRNAs to the Trim32-Ago complex might be a consequence of which Ago is bound to Trim32, or to the ability of Trim32 to alter the properties of the miRISC.
mLin41
Based on mouse and human genetics, as discussed above, Trim2 and Trim32 are developmental regulators of the mammalian CNS. Mouse LIN-41 (mLin41) is the third mammalian Trim-NHL protein shown by genetic means to be involved in CNS morphogenesis.97 Homozygous disruption of mLin41 in gene-trap lines revealed that mLin41 is required for embryonic viability, with a striking but uncharacterized failure of neural tube closure. Using the gene-trap as a reporter for mLin41 promoter activity in heterozygotes, strong expression was detected in the brain, dorsal root ganglia, eyes and branchial arches (and elsewhere) between E10.5 and E12.5. This corresponds closely to the period of embryonic demise in the homozygotes.97 However, it is unclear if the craniofacial abnormalities and closure defect is specific or if it is a secondary result of growth arrest or reduced embryonic viability. Probing mLin41 expression at the protein level, we confirmed mLin41 expression in embryonic stem cells and in Oct-4+ cells of the E7 embryonic ectoderm.23 After embryonic induction of let-7 and miR-125 combine to downregulate mLin41, postnatal niches of mLin41 expression were found in ciliated epithelia of the male and female reproductive tract, in male germ cells and in interfollicular epidermal stem cells. Löer et al had previously demonstrated the mLin41 protein in muscle53 and, thus, the expression pattern of mLin41 seems quite analogous to that of the C. elegans protein.4
In pluripotent cells and in transfection assays in heterologous cells mLin41 was found to accumulate in cytoplasmic P-bodies, as confirmed by colocalization with the P-body markers Dcp1a and Hedls as well as the miRISC proteins Ago2, Mov10 and Tnrc6b.23 This observation dovetailed with a report that the C. elegans protein copurifies with the Dicer protein.98 Association with Dicer was confirmed for mLin41 and extended to Ago2.23 The interaction surface was mapped to the coiled-coil domain, suggesting the NHL domain is free to participate in additional protein-protein interactions. Although the association with Ago2 was studied in greater detail, cotransfection assays with tagged proteins confirmed interaction with Ago1 and Ago4. mLin41 is thus the sixth Trim-NHL protein shown to interact with one of the Argonautes (Mei-P26, Brat, Dappled,65 NHL-282 and Trim3237). Unlike the others, however, mLin41 was shown to mediate degradative ubiquitination of Ago2.23 First, in an autoubiquitination assay E3 ubiquitin ligase activity was confirmed, with the same E2 preference displayed by Trim2 (UbcH5a).22 Adding immunoprecipitated Ago2 to the assay led to the accumulation of polyubiquitinated Ago2. In a transfection assay, Ago2 ubiquitination was dependent on an intact RING domain and was strongly enhanced in the presence of the proteasome inhibitor MG132. In keeping with these results, depletion of endogenous mLin41 in embryocarcinoma cells decreased Ago2 ubiquitination and increased steady state Ago2 levels.23
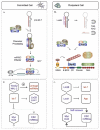
If mLin41 regulates Ago2 turnover and potentially other miRISC components, it should also act as a general inhibitor of miRNA-mediated silencing. Two observations support such a role. Argonautes are thought to be limiting for miRNA-mediated silencing and ectopic Ago2 expression enhances miRNA accumulation,99 most likely by protecting small RNAs from ribonucleolytic degradation.100 mLin41 was shown to block the ability of Ago2 to increase let-7 levels.23 It will be interesting to see if a similar mechanism is responsible for the global reduction in miRNA levels observed after overexpression of Mei-P26.65 Furthermore, mLin41 reduced silencing mediated by let-7, miR-124 or miR-128 in reporter assays. Interference with miRNA activity was dependent on an intact RING domain and could be compensated by increased expression of Ago2.23 This finding was expanded by demonstrating that mLin41 cooperated with Lin28 in suppressing let-7 in the reporter assay, providing evidence for dual negative autoregulatory loops in the post-transcriptional control of let-7 in pluripotent cells (see Fig. 4a and 4b for a schematic view).
Conclusion
Trim-NHL proteins serve as pleiotropic regulators of developmental processes and cell function. In development, a picture is emerging in which Trim-NHL proteins coordinate miRNA activity to sequentially drive transitions between self-renewal, commitment and terminal differentiation of stem cells. The picture is incomplete, in part because recent advances have come from parallel studies in C. elegans, D. melanogaster and mammals so that the issues of sequential action and redundancy have not yet been thoroughly addressed for any one developmental context. For example, in C. elegans LIN-41 and NHL-2 have opposite roles in the heterochronic pathway governing stem cell maturation in the hypodermis and vulva.82 Assuming that LIN-41 is an inhibitor of miRNA pathway activity, as has been shown for the mouse protein, then LIN-41 most likely blocks the acquisition of adult cell fates in the hypodermis at least in part by inhibiting full let-7 activity (the primary miRNA driver of maturation in progenitor populations). Since LIN-41 is itself a let-7 target,4 this is yet another example of a double negative loop enhancing the potency of a miRNA-target gene interaction (see Fig. 4). By promoting miRNA activity, NHL-2 can flip this switch and unleash the full effects of let-7 on downstream heterochronic regulators.
Specification of mouse embryonic stem cells (ESCs) appears to proceed by a similar mechanism. In undifferentiated, pluripotent cells self-renewal is reinforced by a regulatory loop comprising a unique class of ESC-specific miRNAs and transcription factors such as Oct-4, Nanog and Myc.91,101 In ESCs, let-7 is inhibited by the combined action of Lin28 and Lin4123 (reviewed in ref. 102 and in Chapter 7). The importance of let-7 is threefold. First, the impact of let-7 on the proteome is amplified due to direct targeting of miRNA pathway genes.23,96,103-105 Second, let-7 targets cell cycle regulators, thereby directly influencing proliferation.106 Third, let-7 suppresses the transcriptional program of transcription factors required for the maintenance of pluripotency and self-renewal by both direct (c-Myc, N-Myc) and indirect mechanisms (Oct-4, Nanog, Sox2, Tcf3).91 By analogy to the opposing action of LIN-41 and NHL-2 in C. elegans, it is reasonable to expect a counterpoint to mLin41 that activates the miRNA pathway during commitment. In neural lineages Trim32 fulfills two of the expected criteria: it activates a select class of miRNAs, including let-7a and suppresses c-Myc.37
The dual function exerted by at least some Trim-NHLs in the ubiquitin and miRNA pathways should provide a framework for exploring their functional pleiotropy. It will be important to determine whether miRISC association is common to all family members. Within the miRISC, evidence from a variety of sources suggests that Ago2 is not the only substrate for ubiquitination.23,107,108 The ubiquitination of additional miRISC components could certainly affect more than just the turnover rate of the individual proteins in the complex. Complex assembly, activity and intracellular compartmentalization might also be influenced. One obvious possibility is that the ability of many Trim-NHLs to associate with cytoskeletal components in a variety of cellular compartments may reflect a role in miRISC transport.108-111 Alternatively, the association of Wech with focal adhesions or Trim3 with CART may be evidence of miRISC independent activities.
A recent study directly assayed functional redundancy and cooperation among the five Trim-NHLs in C. elegans. Mutations in four of the five (with the exception of LIN-41) were able to suppress a mutation in the embryonic polarity gene par-2, suggesting substantial functional redundancy in this pathway.10 However, other specific functions differed10 and the function of NHL-2 in the heterochronic pathway later in development appears to be independent of NCL-1, NHL-1 or NHL-3.82 In the mouse, loss of Lin41 cannot be compensated and leads to embryonic lethality.97 On the other hand, no obvious defect in neurogenesis was reported after deletion of Trim32,44 in contrast to the direct assays of neuronal differentiation reported by Schwamborn et al.37 In this case Trim2 and Trim3 may functionally compensate for Trim32 deficiency. Coiled-coil domains can mediate heteromeric interactions,9 but there is no evidence yet for cooperativity among Trim-NHLs or between Trim-NHLs and other E3 ligases. If Trim-NHLs can form heterodimers, then the RING-less proteins Brat and Dappled/Wech might promote ubiquitination indirectly.
The discovery of ubiquitin-mediated regulation of miRNAs has revealed a nexus between two of the cells' most powerful post-transcriptional regulatory pathways. The miRNA pathway appears to make extensive use of autoregulatory feedback loops to adjust miRNA activity during development and stem cell differentiation. Several of the key molecules in these loops are involved in tumor formation (Brat in Drosophila or let-7, Lin28 and Myc in humans), underscoring their potential relevance for human disease. Two developmental disturbances have been linked to Trim32, more may be uncovered as the other human Trim-NHLs are studied. In addition to development, regulation of Trim-NHL protein synthesis or activity, for example in response to cell signaling, may allow cells to modulate miRNA efficiency to maintain cellular homeostasis.
Acknowledgements
F.G.W., E.F. and A.R. received support from the DFG Collaborative research project 665; E.C. and A.R were supported by the DFG Graduate School 1123.
References
- 1.
- Reinhart BJ, Slack FJ, Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403(6772):901–906. [PubMed: 10706289]
- 2.
- Pasquinelli AE, Reinhart BJ, Slack F, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408(6808):86–89. [PubMed: 11081512]
- 3.
- Lee R, Feinbaum R, Ambros V. A short history of a short RNA. Cell. 2004;116(2 Suppl):S89-92–81. [PubMed: 15055592]
- 4.
- Slack FJ, Basson M, Liu Z, et al. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell. 2000;5(4):659–669. [PubMed: 10882102]
- 5.
- Slack FJ, Ruvkun G. A novel repeat domain that is often associated with RING finger and B-box motifs. Trends Biochem Sci. 1998;23(12):474–475. [PubMed: 9868369]
- 6.
- Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of 'single protein RING finger' E3 ubiquitin ligases. Bioessays. 2005;27(11):1147–1157. [PubMed: 16237670]
- 7.
- Sardiello M, Cairo S, Fontanella B, et al. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol Biol. 2008;8:225. [PMC free article: PMC2533329] [PubMed: 18673550]
- 8.
- Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005;3(10):799–808. [PubMed: 16175175]
- 9.
- Reymond A, Meroni G, Fantozzi A, et al. The tripartite motif family identifies cell compartments. Embo J. 2001;20(9):2140–2151. [PMC free article: PMC125245] [PubMed: 11331580]
- 10.
- Hyenne V, Desrosiers M, Labbe JC. C. elegans Brat homologs regulate PAR protein-dependent polarity and asymmetric cell division. Dev Biol. 2008;321(2):368–378. [PubMed: 18652816]
- 11.
- O'Farrell F, Esfahani SS, Engstrom Y, et al. Regulation of the Drosophila lin-41 homologue dappled by let-7 reveals conservation of a regulatory mechanism within the LIN-41 subclade. Dev Dyn. 2008;237(1):196–208. [PubMed: 18069688]
- 12.
- Saurin AJ, Borden KL, Boddy MN, et al. Does this have a familiar RING? Trends Biochem Sci. 1996;21(6):208–214. [PubMed: 8744354]
- 13.
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. [PubMed: 19489725]
- 14.
- Joazeiro CA, Wing SS, Huang H, et al. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999;286(5438):309–312. [PubMed: 10514377]
- 15.
- Yokouchi M, Kondo T, Houghton A, et al. Ligand-induced ubiquitination of the epidermal growth factor receptor involves the interaction of the c-Cbl RING finger and UbcH7. J Biol Chem. 1999;274(44):31707–31712. [PubMed: 10531381]
- 16.
- Lorick KL, Jensen JP, Fang S, et al. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA. 1999;96(20):11364–11369. [PMC free article: PMC18039] [PubMed: 10500182]
- 17.
- Freemont PS. RING for destruction? Curr Biol. 2000;10(2):R84–87. [PubMed: 10662664]
- 18.
- Lupas A. Coiled coils: new structures and new functions. Trends Biochem Sci. 1996;21(10):375–382. [PubMed: 8918191]
- 19.
- Zheng N, Wang P, Jeffrey PD, et al. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102(4):533–539. [PubMed: 10966114]
- 20.
- Horn EJ, Albor A, Liu Y, et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis. 2004;25(2):157–167. [PubMed: 14578165]
- 21.
- Kudryashova E, Kudryashov D, Kramerova I, et al. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J Mol Biol. 2005;354(2):413–424. [PubMed: 16243356]
- 22.
- Balastik M, Ferraguti F, Pires-da Silva A, et al. Deficiency in ubiquitin ligase TRIM2 causes accumulation of neurofilament light chain and neurodegeneration. Proc Natl Acad Sci USA. 2008;105(33):12016–12021. [PMC free article: PMC2575299] [PubMed: 18687884]
- 23.
- Rybak A, Fuchs H, Hadian K, et al. The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat Cell Biol. 2009;11(12):1411–1420. [PubMed: 19898466]
- 24.
- Chiang AP, Beck JS, Yen HJ, et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc Natl Acad Sci USA. 2006;103(16):6287–6292. [PMC free article: PMC1458870] [PubMed: 16606853]
- 25.
- Saccone V, Palmieri M, Passamano L, et al. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum Mutat. 2008;29(2):240–247. [PubMed: 17994549]
- 26.
- Massiah MA, Matts JA, Short KM, et al. Solution structure of the MID1 B-box2 CHC(D/C)C(2)H(2) zinc-binding domain: insights into an evolutionarily conserved RING fold. J Mol Biol. 2007;369(1):1–10. [PubMed: 17428496]
- 27.
- Massiah MA, Simmons BN, Short KM, et al. Solution structure of the RBCC/TRIM B-box1 domain of human MID1: B-box with a RING. J Mol Biol. 2006;358(2):532–545. [PubMed: 16529770]
- 28.
- Tao H, Simmons BN, Singireddy S, et al. Structure of the MID1 tandem B-boxes reveals an interaction reminiscent of intermolecular ring heterodimers. Biochemistry. 2008;47(8):2450–2457. [PubMed: 18220417]
- 29.
- Grigoryan G, Keating AE. Structural specificity in coiled-coil interactions. Curr Opin Struct Biol. 2008;18(4):477–483. [PMC free article: PMC2567808] [PubMed: 18555680]
- 30.
- Zhou AX, Hartwig JH, Akyurek LM. Trends Cell Biol. Filamins in cell signaling, transcription and organ development. [PubMed: 20061151]
- 31.
- Arama E, Dickman D, Kimchie Z, et al. Mutations in the beta-propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene. 2000;19(33):3706–3716. [PubMed: 10949924]
- 32.
- Sonoda J, Wharton RP. Drosophila brain tumor is a translational repressor. Genes Dev. 2001;15(6):762–773. [PMC free article: PMC312658] [PubMed: 11274060]
- 33.
- Edwards TA, Wilkinson BD, Wharton RP, et al. Model of the brain tumor-Pumilio translation repressor complex. Genes Dev. 2003;17(20):2508–2513. [PMC free article: PMC218144] [PubMed: 14561773]
- 34.
- Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5(3):177–187. [PubMed: 14990998]
- 35.
- Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10(5):319–331. [PMC free article: PMC2712597] [PubMed: 19352404]
- 36.
- Albor A, El-Hizawi S, Horn EJ, et al. The interaction of Piasy with Trim32, an E3-ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2H, promotes Piasy degradation and regulates UVB-induced keratinocyte apoptosis through NFkappaB. J Biol Chem. 2006;281(35):25850–25866. [PubMed: 16816390]
- 37.
- Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136(5):913–925. [PMC free article: PMC2988196] [PubMed: 19269368]
- 38.
- Locke M, Tinsley CL, Benson MA, et al. TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum Mol Genet. 2009;18(13):2344–2358. [PMC free article: PMC2694686] [PubMed: 19349376]
- 39.
- Fridell RA, Harding LS, Bogerd HP, et al. Identification of a novel human zinc finger protein that specifically interacts with the activation domain of lentiviral Tat proteins. Virology. 1995;209(2):347–357. [PubMed: 7778269]
- 40.
- Frosk P, Weiler T, Nylen E, et al. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am J Hum Genet. 2002;70(3):663–672. [PMC free article: PMC447621] [PubMed: 11822024]
- 41.
- Li W, Zhang Q, Oiso N, et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet. 2003;35(1):84–89. [PMC free article: PMC2860733] [PubMed: 12923531]
- 42.
- Mykytyn K, Sheffield VC. Establishing a connection between cilia and Bardet-Biedl Syndrome. Trends Mol Med. 2004;10(3):106–109. [PubMed: 15106604]
- 43.
- Ansley SJ, Badano JL, Blacque OE, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425(6958):628–633. [PubMed: 14520415]
- 44.
- Kudryashova E, Wu J, Havton LA, et al. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum Mol Genet. 2009;18(7):1353–1367. [PMC free article: PMC2722196] [PubMed: 19155210]
- 45.
- Pasquinelli AE, Ruvkun G. Control of developmental timing by micrornas and their targets. Annu Rev Cell Dev Biol. 2002;18:495–513. [PubMed: 12142272]
- 46.
- Kloosterman WP, Wienholds E, Ketting RF, et al. Substrate requirements for let-7 function in the developing zebrafish embryo. Nucleic Acids Res. 2004;32(21):6284–6291. [PMC free article: PMC535676] [PubMed: 15585662]
- 47.
- Lancman JJ, Caruccio NC, Harfe BD, et al. Analysis of the regulation of lin-41 during chick and mouse limb development. Dev Dyn. 2005;234(4):948–960. [PubMed: 16245339]
- 48.
- Schulman BR, Esquela-Kerscher A, Slack FJ. Reciprocal expression of lin-41 and the microRNAs let-7 and mir-125 during mouse embryogenesis. Dev Dyn. 2005;234(4):1046–1054. [PMC free article: PMC2596717] [PubMed: 16247770]
- 49.
- Kanamoto T, Terada K, Yoshikawa H, et al. Cloning and regulation of the vertebrate homologue of lin-41 that functions as a heterochronic gene in Caenorhabditis elegans. Dev Dyn. 2006;235(4):1142–1149. [PubMed: 16477647]
- 50.
- Rodriguez A, Zhou Z, Tang ML, et al. Identification of immune system and response genes and novel mutations causing melanotic tumor formation in Drosophila melanogaster. Genetics. 1996;143(2):929–940. [PMC free article: PMC1207349] [PubMed: 8725239]
- 51.
- Brody T, Stivers C, Nagle J, et al. Identification of novel Drosophila neural precursor genes using a differential embryonic head cDNA screen. Mech Dev. 2002;113(1):41–59. [PubMed: 11900973]
- 52.
- O'Farrell F, Kylsten P. A mis-expression study of factors affecting Drosophila PNS cell identity. Biochem Biophys Res Commun. 2008;370(4):657–662. [PubMed: 18420029]
- 53.
- Loer B, Bauer R, Bornheim R, et al. The NHL-domain protein Wech is crucial for the integrin-cytoskeleton link. Nat Cell Biol. 2008;10(4):422–428. [PubMed: 18327251]
- 54.
- Delon I, Brown N. Cell-matrix adhesion: the wech connection. Curr Biol. 2008;18(9):R389–391. [PubMed: 18460322]
- 55.
- Loer B, Hoch M. Wech proteins: roles in integrin functions and beyond. Cell Adh Migr. 2008;2(3):177–179. [PMC free article: PMC2634098] [PubMed: 19262117]
- 56.
- Sokol NS, Xu P, Jan YN, et al. Drosophila let-7 microRNA is required for remodeling of the neuromusculature during metamorphosis. Genes Dev. 2008;22(12):1591–1596. [PMC free article: PMC2428057] [PubMed: 18559475]
- 57.
- El Husseini AE, Vincent SR. Cloning and characterization of a novel RING finger protein that interacts with class V myosins. J Biol Chem. 1999;274(28):19771–19777. [PubMed: 10391919]
- 58.
- Trybus KM. Myosin V from head to tail. Cell Mol Life Sci. 2008;65(9):1378–1389. [PMC free article: PMC2613318] [PubMed: 18239852]
- 59.
- Yan Q, Sun W, Kujala P, et al. CART: an Hrs/actinin-4/BERP/myosin V protein complex required for efficient receptor recycling. Mol Biol Cell. 2005;16(5):2470–2482. [PMC free article: PMC1087250] [PubMed: 15772161]
- 60.
- Mosesson Y, Chetrit D, Schley L, et al. Monoubiquitinylation regulates endosomal localization of Lst2, a negative regulator of EGF receptor signaling. Dev Cell. 2009;16(5):687–698. [PMC free article: PMC3356849] [PubMed: 19460345]
- 61.
- Zwang Y, Yarden Y. Systems biology of growth factor-induced receptor endocytosis. Traffic. 2009;10(4):349–363. [PubMed: 19183301]
- 62.
- Ohkawa N, Kokura K, Matsu-Ura T, et al. Molecular cloning and characterization of neural activity-related RING finger protein (NARF): a new member of the RBCC family is a candidate for the partner of myosin V. J Neurochem. 2001;78(1):75–87. [PubMed: 11432975]
- 63.
- Gray PA, Fu H, Luo P, et al. Mouse brain organization revealed through direct genome-scale TF expression analysis. Science. 2004;306(5705):2255–2257. [PubMed: 15618518]
- 64.
- Wright TR. The Wilhelmine E. Key 1992 Invitational lecture. Phenotypic analysis of the Dopa decarboxylase gene cluster mutants in Drosophila melanogaster. J Hered. 1996;87(3):175–190. [PubMed: 8683095]
- 65.
- Neumuller RA, Betschinger J, Fischer A, et al. Mei-P26 regulates microRNAs and cell growth in the Drosophila ovarian stem cell lineage. Nature. 2008;454(7201):241–245. [PMC free article: PMC2988194] [PubMed: 18528333]
- 66.
- Frank DJ, Edgar BA, Roth MB. The Drosophila melanogaster gene brain tumor negatively regulates cell growth and ribosomal RNA synthesis. Development. 2002;129(2):399–407. [PubMed: 11807032]
- 67.
- Bello B, Reichert H, Hirth F. The brain tumor gene negatively regulates neural progenitor cell proliferation in the larval central brain of Drosophila. Development. 2006;133(14):2639–2648. [PubMed: 16774999]
- 68.
- Bowman SK, Rolland V, Betschinger J, et al. The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev Cell. 2008;14(4):535–546. [PMC free article: PMC2988195] [PubMed: 18342578]
- 69.
- Knoblich JA. Mechanisms of asymmetric stem cell division. Cell. 2008;132(4):583–597. [PubMed: 18295577]
- 70.
- Kohlmaier A, Edgar BA. Proliferative control in Drosophila stem cells. Curr Opin Cell Biol. 2008;20(6):699–706. [PMC free article: PMC2650021] [PubMed: 18996190]
- 71.
- Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124(6):1241–1253. [PubMed: 16564014]
- 72.
- Lee CY, Wilkinson BD, Siegrist SE, et al. Brat is a Miranda cargo protein that promotes neuronal differentiation and inhibits neuroblast self-renewal. Dev Cell. 2006;10(4):441–449. [PubMed: 16549393]
- 73.
- Sekelsky JJ, McKim KS, Messina L, et al. Identification of novel Drosophila meiotic genes recovered in a P-element screen. Genetics. 1999;152(2):529–542. [PMC free article: PMC1460643] [PubMed: 10353897]
- 74.
- Page SL, McKim KS, Deneen B, et al. Genetic studies of mei-P26 reveal a link between the processes that control germ cell proliferation in both sexes and those that control meiotic exchange in Drosophila. Genetics. 2000;155(4):1757–1772. [PMC free article: PMC1461182] [PubMed: 10924472]
- 75.
- Ivanov AI, Rovescalli AC, Pozzi P, et al. Genes required for Drosophila nervous system development identified by RNA interference. Proc Natl Acad Sci USA. 2004;101(46):16216–16221. [PMC free article: PMC528945] [PubMed: 15534205]
- 76.
- Glasscock E, Singhania A, Tanouye MA. The mei-P26 gene encodes a RING finger B-box coiled-coil-NHL protein that regulates seizure susceptibility in Drosophilia. Genetics. 2005;170(4):1677–1689. [PMC free article: PMC1449765] [PubMed: 15937125]
- 77.
- Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure and paralysis. Cell. 1994;79(1):23–33. [PubMed: 7923374]
- 78.
- Pascual A, Chaminade M, Preat T. Ethanolamine kinase controls neuroblast divisions in Drosophila mushroom bodies. Dev Biol. 2005;280(1):177–186. [PubMed: 15766757]
- 79.
- Okamura K, Ishizuka A, Siomi H, et al. Distinct roles for argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev. 2004;18(14):1655–1666. [PMC free article: PMC478188] [PubMed: 15231716]
- 80.
- Park JK, Liu X, Strauss TJ, et al. The miRNA pathway intrinsically controls self-renewal of Drosophila germline stem cells. Curr Biol. 2007;17(6):533–538. [PubMed: 17320391]
- 81.
- Jin Z, Xie T. Dcr-1 maintains Drosophila ovarian stem cells. Curr Biol. 2007;17(6):539–544. [PubMed: 17306537]
- 82.
- Hammell CM, Lubin I, Boag PR, et al. nhl-2 Modulates microRNA activity in Caenorhabditis elegans. Cell. 2009;136(5):926–938. [PMC free article: PMC2670343] [PubMed: 19269369]
- 83.
- Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006;4(7):e210. [PMC free article: PMC1475773] [PubMed: 16756390]
- 84.
- Loedige I, Filipowicz W. TRIM-NHL proteins take on miRNA regulation. Cell. 2009;136(5):818–820. [PubMed: 19269362]
- 85.
- Coller JM, Tucker M, Sheth U, et al. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA. 2001;7(12):1717–1727. [PMC free article: PMC1370212] [PubMed: 11780629]
- 86.
- Eulalio A, Rehwinkel J, Stricker M, et al. Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing. Genes Dev. 2007;21(20):2558–2570. [PMC free article: PMC2000321] [PubMed: 17901217]
- 87.
- Farkas LM, Huttner WB. The cell biology of neural stem and progenitor cells and its significance for their proliferation versus differentiation during mammalian brain development. Curr Opin Cell Biol. 2008;20(6):707–715. [PubMed: 18930817]
- 88.
- Zhong W, Chia W. Neurogenesis and asymmetric cell division. Curr Opin Neurobiol. 2008;18(1):4–11. [PubMed: 18513950]
- 89.
- Kosodo Y, Huttner WB. Basal process and cell divisions of neural progenitors in the developing brain. Dev Growth Differ. 2009;51(3):251–261. [PubMed: 19379277]
- 90.
- Bussing I, Slack FJ. Grosshans H. let-7 microRNAs in development, stem cells and cancer. Trends Mol Med. 2008;14(9):400–409. [PubMed: 18674967]
- 91.
- Melton C, Judson RL, Blelloch R. Nature. 2010. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. [PMC free article: PMC2894702] [PubMed: 20054295]
- 92.
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16(20):2699–2712. [PMC free article: PMC187459] [PubMed: 12381668]
- 93.
- Martins RA, Zindy F, Donovan S, et al. N-myc coordinates retinal growth with eye size during mouse development. Genes Dev. 2008;22(2):179–193. [PMC free article: PMC2192753] [PubMed: 18198336]
- 94.
- Kloosterman WP, Wienholds E, de Bruijn E, et al. In situ detection of miRNAs in animal embryos using LNA-modified oligonucleotide probes. Nat Methods. 2006;3(1):27–29. [PubMed: 16369549]
- 95.
- Nishino J, Kim I, Chada K, et al. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135(2):227–239. [PMC free article: PMC2582221] [PubMed: 18957199]
- 96.
- Rybak A, Fuchs H, Smirnova L, et al. A feedback loop comprising lin-28 and let-7 controls prelet-7 maturation during neural stem-cell commitment. Nat Cell Biol. 2008;10(8):987–993. [PubMed: 18604195]
- 97.
- Maller Schulman BR, Liang X, Stahlhut C, et al. The let-7 microRNA target gene, Mlin41/Trim71 is required for mouse embryonic survival and neural tube closure. Cell Cycle. 2008;7(24):3935–3942. [PMC free article: PMC2895810] [PubMed: 19098426]
- 98.
- Duchaine TF, Wohlschlegel JA, Kennedy S, et al. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell. 2006;124(2):343–354. [PubMed: 16439208]
- 99.
- Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and post-transcriptional regulation of microRNA expression. Cell. 2007;131(6):1097–1108. [PubMed: 18083100]
- 100.
- Chatterjee S, Grosshans H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature. 2009;461(7263):546–549. [PubMed: 19734881]
- 101.
- Sinkkonen L, Hugenschmidt T, Berninger P, et al. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 2008;15(3):259–267. [PubMed: 18311153]
- 102.
- Dueck A, Meister G. TRIMming microRNA function in mouse stem cells. Nat Cell Biol. 2009;11(12):1392–1393. [PubMed: 19949436]
- 103.
- Selbach M, Schwanhausser B, Thierfelder N, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455(7209):58–63. [PubMed: 18668040]
- 104.
- Ding XC, Slack FJ, Grosshans H. The let-7 microRNA interfaces extensively with the translation machinery to regulate cell differentiation. Cell Cycle. 2008;7(19):3083–3090. [PMC free article: PMC2887667] [PubMed: 18818519]
- 105.
- Tokumaru S, Suzuki M, Yamada H, et al. Carcinogenesis. 2008. let-7 regulates Dicer expression and constitutes a negative feedback loop. [PubMed: 18700235]
- 106.
- Johnson CD, Esquela-Kerscher A, Stefani G, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67(16):7713–7722. [PubMed: 17699775]
- 107.
- Ashraf SI, McLoon AL, Sclarsic SM, et al. Synaptic protein synthesis associated with memory is regulated by the RISC pathway in Drosophila. Cell. 2006;124(1):191–205. [PubMed: 16413491]
- 108.
- Gibbings DJ, Ciaudo C, Erhardt M, et al. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. 2009;11(9):1143–1149. [PubMed: 19684575]
- 109.
- Kotaja N, Bhattacharyya SN, Jaskiewicz L, et al. The chromatoid body of male germ cells: Similarity with processing bodies and presence of Dicer and microRNA pathway components. Proc Natl Acad Sci USA. 2006 [PMC free article: PMC1413789] [PubMed: 16477042]
- 110.
- Wulczyn FG, Smirnova L, Rybak A, et al. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. Faseb J. 2007;21(2):415–426. [PubMed: 17167072]
- 111.
- Lee YS, Pressman S, Andress AP, et al. Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol. 2009;11(9):1150–1156. [PMC free article: PMC2737091] [PubMed: 19684574]
- 112.
- Kirilly D, Xie T. The Drosophila ovary: an active stem cell community. Cell Res. 2007;17(1):15–25. [PubMed: 17199109]
- miRNAs need a Trim Regulation of miRNA Activity by Trim-NHL Proteins - Madame Cu...miRNAs need a Trim Regulation of miRNA Activity by Trim-NHL Proteins - Madame Curie Bioscience Database
- GSK3-Signal Regulation of Pattern Formation in Dictyostelium: Wnt-Like Pathways ...GSK3-Signal Regulation of Pattern Formation in Dictyostelium: Wnt-Like Pathways During Non-Canonical Multicellular Development - Madame Curie Bioscience Database
- A New Member of the CtBP/BARS Family from Plants: Angustifolia - Madame Curie Bi...A New Member of the CtBP/BARS Family from Plants: Angustifolia - Madame Curie Bioscience Database
- Shh Expression in Pulmonary Injury and Disease - Madame Curie Bioscience Databas...Shh Expression in Pulmonary Injury and Disease - Madame Curie Bioscience Database
- Preimplantation Genetic Diagnosis (PGD): Screening for Aneuploidy in Human Oocyt...Preimplantation Genetic Diagnosis (PGD): Screening for Aneuploidy in Human Oocytes and Polar Bodies - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...