Summary
Clinical characteristics.
Angelman syndrome (AS) is characterized by severe developmental delay or intellectual disability, severe speech impairment, gait ataxia and/or tremulousness of the limbs, and unique behavior with an apparent happy demeanor that includes frequent laughing, smiling, and excitability. Microcephaly and seizures are also common. Developmental delays are first noted at around age six months; however, the unique clinical features of AS do not become manifest until after age one year.
Diagnosis/testing.
The diagnosis of AS is established in a proband who meets the consensus clinical diagnostic criteria and/or who has findings on molecular genetic testing that suggest deficient expression or function of the maternally inherited UBE3A allele. Analysis of parent-specific DNA methylation imprints in the 15q11.2-q13 chromosome region detects approximately 80% of individuals with AS, including those with a deletion, uniparental disomy, or an imprinting defect; fewer than 1% of individuals have a cytogenetically visible chromosome rearrangement (e.g., translocation or inversion). UBE3A sequence analysis detects pathogenic variants in an additional approximately 11% of individuals. Therefore, molecular genetic testing (methylation analysis and UBE3A sequence analysis) identifies alterations in approximately 90% of individuals. The remaining 10% of individuals with classic phenotypic features of AS have the disorder as a result of an as-yet unidentified genetic mechanism.
Management.
Treatment of manifestations: Anti-seizure medication for seizures. Accommodation for hypermotoric behaviors and disruptive nighttime wakefulness. Behavior modification can be effective for disruptive or self-injurious behaviors. Physical therapy, occupational therapy, and speech therapy with an emphasis on nonverbal methods of communication, including augmentative communication aids (e.g., picture cards, communication boards) and signing. Individualization and flexibility in school settings. Routine management of gastroesophageal reflux, feeding difficulties, constipation, and strabismus. Thoraco-lumbar jackets and/or surgical intervention for scoliosis. Bracing or surgery as needed for subluxed or pronated ankles or tight Achilles tendons.
Surveillance: Monitor for new seizures and/or changes in seizures, developmental progress, behavior issues, mobility, motor skills, gastroesophageal reflux, constipation, and feeding issues at each visit. Evaluation of older children for obesity associated with an excessive appetite. Annual clinical examination for scoliosis; ophthalmology examination in the first year if strabismus is present; ophthalmology exam at age two years with follow up per ophthalmologist; clinical examination for scoliosis annually.
Agents/circumstances to avoid: Overtreatment with sedating medications in order to reduce hyperexcitable and hypermotoric behavior. Overtreatment with anti-seizure medication when movement abnormalities are mistaken for seizures and/or when EEG abnormalities persist even as seizures are controlled.
Genetic counseling.
Individuals with AS typically represent simplex cases (i.e., a single affected family member) and have the disorder as the result of a de novo genetic alteration associated with a very low recurrence risk. Less commonly, an individual with AS has the disorder as the result of a genetic alteration associated with an imprinting pattern of autosomal dominant inheritance or variable recurrence risk. Reliable recurrence risk assessment therefore requires identification of the underlying genetic mechanism in the proband and confirmation of the genetic status of the parents. Prenatal detection of all the known molecular genetic alterations in the 15q11.2-q13 region that give rise to AS is possible and is an option for families once the underlying genetic mechanism in the proband has been identified.
Diagnosis
Consensus criteria for the clinical diagnosis of Angelman syndrome (AS) have been developed in conjunction with the Scientific Advisory Committee of the US Angelman Syndrome Foundation [Williams et al 2006]. Several reviews are available [Bird 2014, Buiting et al 2016, Prasad et al 2018].
Suggestive Findings
AS should be suspected in individuals with the following clinical, laboratory, and radiographic findings.
Clinical
- Normal prenatal and birth history, normal head circumference at birth, no major birth defects
- Delayed attainment of developmental milestones by age six to 12 months, eventually classified as severe, without loss of skills
- Speech impairment, with minimal to no use of words; receptive language skills and nonverbal communication skills higher than expressive language skills
- Movement or balance disorder, usually ataxia of gait and/or tremulous movement of the limbs
- Behavioral uniqueness including any combination of frequent laughter/smiling, apparent happy demeanor, excitability (often with hand-flapping movements), and hypermotoric behavior
Clinical criteria that help establish the diagnosis [Williams et al 2006]
Findings in more than 80% of affected individuals
- Delayed or disproportionately slow growth in head circumference, usually resulting in absolute or relative microcephaly by age two years
- Seizures, usually starting before age three years
- Abnormal EEG, with a characteristic pattern of large-amplitude slow-spike waves
Findings in fewer than 80% of affected individuals
- Craniofacial features including flat occiput, occipital groove, wide mouth, widely spaced teeth, protruding tongue, prognathia (See Figure 1.)
- Feeding problems and/or hypotonia during infancy, tongue thrusting, suck/swallowing disorders, frequent drooling, excessive chewing/mouthing behaviors
- Strabismus
- Hypopigmented skin, light hair and eye color compared to family members; seen only in those with a 15q11.2-q13 deletion
- Hyperactive lower-extremity deep-tendon reflexes
- Uplifted, flexed arm position especially during ambulation
- Wide-based gait with pronated or valgus-positioned ankles
- Increased sensitivity to heat
- Abnormal sleep-wake cycles and diminished need for sleep
- Attraction to and fascination with water; fascination with crinkly items such as certain papers and plastics
- Abnormal food-related behaviors
- Obesity (in the older child; more common in those who do not have a 15q11.2-q13 deletion)
- Scoliosis
- Constipation

Laboratory
Deletion of the 15q11.2-q13 genomic region (detected by chromosomal microarray or other methods) is suggestive of AS but not, in and of itself, diagnostic.
Metabolic, hematologic, and chemical laboratory profiles are normal.
Radiographic
Brain imaging shows structurally normal brain by MRI or CT, although mild cortical atrophy or dysmyelination may be observed.
Establishing the Diagnosis
The clinical diagnosis of AS can be established in a proband based on clinical diagnostic criteria (see Suggestive Findings, Clinical criteria that help establish the diagnosis) [Williams et al 2006] or the molecular diagnosis can be established in a proband with suggestive findings and findings on molecular genetic testing that suggest deficient expression or function of the maternally inherited UBE3A allele (see Table 1).
Molecular diagnosis. The diagnosis of AS is established in a proband with suggestive findings who has one of the following on molecular genetic testing (see Table 1):
- Abnormal methylation at 15q11.2-q13 due to one of the following:
- Deletion of the maternally inherited 15q11.2-q13 region (which includes UBE3A)
- Uniparental disomy (UPD) of the paternal chromosome region 15q11.2-q13
- An imprinting defect of the maternal chromosome 15q11.2-q13 region
- A pathogenic variant in the maternally derived UBE3A
Molecular genetic testing approaches to establish the diagnosis can be based on either the clinical findings or the laboratory findings that suggested the diagnosis of AS.
Based on clinical findings in a symptomatic individual who has not had any prior molecular genetic testing:
- DNA methylation analysis is typically the first test ordered. Individuals with AS caused by a 5- to 7-Mb deletion of 15q11.2-q13, UPD, or an imprinting defect have only an unmethylated (i.e., "paternal") contribution (i.e., an abnormal parent-specific DNA methylation imprint). DNA methylation analysis identifies approximately 80% of individuals with AS.Note: Most commercially available DNA methylation analysis tests cannot distinguish between AS resulting from a 15q11.2-q13 deletion, UPD, or an imprinting defect. Further testing is required to identify the underlying molecular mechanism (see Genetic Counseling).
If DNA methylation analysis is normal:
- Single-gene testing. Sequence analysis of UBE3A is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes UBE3A (e.g., most epilepsy, autism, and intellectual deficiency multigene panels) and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- More comprehensive genomic testing (when available) including exome sequencing, genome sequencing, and mitochondrial sequencing may be considered if DNA methylation and UBE3A analysis (and/or use of a multigene panel) fails to confirm a diagnosis in an individual with features of AS.
Based on laboratory findings in an individual who has been found to have a 15q11.2-q13 deletion on chromosomal microarray (CMA), fluorescent in situ hybridization (FISH), or karyotype,* perform DNA methylation analysis to determine if the deletion is on the maternally derived chromosome 15.
* Fewer than 1% of individuals with AS have a cytogenetically visible chromosome 15 rearrangement (i.e., translocation or inversion) involving 15q11.2-q13.
Table 1.
Molecular Genetic Testing Used in Angelman Syndrome
Possible explanations for the failure to detect AS-causing genetic abnormalities in approximately 10% of individuals with clinically diagnosed AS:
- Incorrect clinical diagnosis
- Undetected pathogenic variants in the regulatory region(s) of UBE3A
- Other unidentified mechanisms or gene(s) involved in UBE3A function
Clinical Characteristics
Clinical Description
Angelman syndrome (AS) is characterized by severe developmental delay and intellectual disability, severe speech impairment, gait ataxia and/or tremulousness of the limbs, and a unique behavior with an apparent happy demeanor that includes frequent laughing, smiling, and excitability. Microcephaly and seizures are also common. Developmental delays are first noted at around age six months; however, the unique clinical features of AS do not become manifest until after age one year.
Table 2.
Angelman Syndrome: Frequency of Select Features
Seizure onset typically occurs between ages one and three years but can occur at any age; most appear by age five years. Epilepsy occurs in up to 90% of individuals and is more commonly observed in those with 15q11.2-q13 deletions [Khan et al 2019, Bindels-de Heus et al 2020]. The seizures are usually associated with generalized, somewhat specific EEG changes: runs of high-amplitude delta activity with intermittent spike and slow-wave discharges (at times observed as a notched delta pattern); runs of rhythmic theta activity over a wide area; and runs of rhythmic sharp theta activity of 5-6/s over the posterior third of the head, forming complexes with small spikes. These are usually facilitated by or seen only with eye closure [Boyd et al 1988, Samanta 2021].
Seizure types can be quite varied; the most common are myoclonic, atonic, generalized tonic-clonic, and atypical absence [Thibert et al 2009, Fiumara et al 2010]. Multiple seizure types occur in up to 50% of individuals. Infantile spasms are rare. Seizures continue to be present throughout adulthood.
Brain MRI may show mild atrophy and mild dysmyelination, but no structural lesions [Harting et al 2009, Castro-Gago et al 2010].
Nonconvulsive status epilepticus (NCSE) may occur in children [Bindels-de Heus et al 2020] and adults [Prasad et al 2018]. This type of status may not be recognized clinically but can be associated with loss of developmental skills and diminished awareness and may last for hours or even days. Most common is an atypical absence or myoclonic type NCSE causing decreased alertness, atypical absence status, atonic head drop, hypotonia, and/or myoclonic movements [Elia 2009, Worden et al 2018].
Nonepileptic myoclonus (NEM), also termed cortical myoclonus, should be distinguished from true seizures that have an EEG signature. NEM can include jerking, tic-like, or twitching movements without obvious alteration in awareness and with no epileptiform EEG changes. NEM typically occurs in teenagers and young adults [Pollack et al 2018].
Sleep problems are common in individuals with AS and include frequent and early awakening, dyssomnias (difficulties initiating or maintaining sleep), fragmented and irregular sleep-wake cycles, disruptive night behaviors such as periods of laughter, and sleep-related seizures [Pelc et al 2008, Spruyt et al 2018]. Sleep difficulties are further influenced by constipation, gastroesophageal reflux disease, and scoliosis [Bindels-de Heus et al 2020]. Sleep related issues may improve with age, though some individuals continue to require co-sleeping [Walz et al 2005, Dosier et al 2017]. Given the comorbidity of behavior issues, seizures, and sleep problems, management largely focuses on behavior modification, epileptic control, medication, and improving sleep hygiene to approach these issues.
Behavioral features include frequent laughter and smiling, apparent happy demeanor, excitability, often with hand-flapping movements, and hypermotoric behavior. Some infants have an apparent happy affect with excessive chortling or paroxysms of laughter. Infants and toddlers may have seemingly ceaseless activity, constantly keeping their hands or toys in their mouth, and/or moving from object to object. After infancy, exploratory play tends to be by oral manipulation and chewing. Essentially all young children with AS have a component of hyperactivity. Males and females appear equally affected. Laughter can be an appropriate response to a humorous situation, but more often appears in response to a nonspecific event (mental or physical stimulus) or possible expression of anxiety. Aggressive and self-injurious behaviors can occur, including pinching, grabbing, biting, slapping, and hitting. These behaviors often represent attention seeking and frustration due to difficulty with communication, rather than ill intent [Arron et al 2011, Sadhwani et al 2019]. Individuals with AS experience the full spectrum of emotions, develop meaningful relationships with family and friends, and participate in household, recreational, and other activities.
Certain behaviors may suggest a diagnosis of autism spectrum disorder (e.g., fascination with water and crinkly items such as certain papers and plastics, increased sensitivity to heat, abnormal food-related behaviors) but social engagement is typically good. Stereotypic behaviors such as lining up of toys or fascination with spinning objects or flashing lights rarely occur. Some individuals with AS have good response to ABA (applied behavior analysis) therapy [Walz & Baranek 2006, Moss & Howlin 2009, Summers 2012].
The behavior profile generally continues into the adult years. Particularly challenging in teenage and adult years include frustration in communicating wants and preferences, seeking sensory stimulation and social attention, and avoidance of undesired situations [Larson et al 2015].
Motor development and tremor. Tremulous movements can be noted prior to age 12 months and are associated with increased deep-tendon reflexes (see also Clinical Description, Nonepileptic myoclonus).
AS may first be suspected in a toddler because of delayed gross motor milestones and hypotonia. Mildly impaired children may walk fairly normally or have minimal toe-walking or prancing gait, at times accompanied by leaning forward. Being placed in a standing position can result in anxiety or rigidity. The average age of walking is between 2.5 and six years [Lossie et al 2001]. In a recent study of 100 children with AS, those with the 15q11.2-q13 deletion achieved walking on average by 58 months and those without the deletion by age 41 months [Bindels-de Heus et al 2020]. Children who are significantly affected have a jerky, robot-like, stiff gait, with legs kept wide based and arms uplifted and flexed with pronated forearms.
Frequently voluntary movements appear irregular. On the mild end they can present as slight jerkiness to uncoordinated coarse movements on the severe end of the spectrum. These movements can prevent reaching for objects, feeding, and walking. Failure to achieve independent walking may be a result of instability resulting from tremor, epilepsy, vision issues, abnormal muscle tone, or balance problems. Ten percent of children are nonambulatory [Clayton-Smith 1993].
Language impairment and cognitive delay are severe. Although formal psychometric testing appears to indicate developmental achievement at around the 24-30 month range, developmental testing is challenging due to language impairment and hypermotoric and attention-deficit behaviors [Peters et al 2004]. Cognitive abilities may be higher than what is captured on testing, but delays are still likely in the severe range. Individuals with the 15q11.2-q13 deletion usually demonstrate the most severe cognitive delays across all domains.
Appropriate and consistent use of one or two words is rare. Babies and young infants have decreased cooing and babbling. At age ten to 18 months, a single word such as "mama" may develop but is often used indiscriminately. In a survey of 47 individuals, 39% spoke up to four words but it was unclear if these words were used with purpose [Buntinx et al 1995]. Larson et al [2015] reported that 13% of individuals had five or more words. Receptive language skills are always more advanced than expressive language skills [Bindels-de Heus et al 2020].
Seizures and significant hyperactivity can impede early communication development including eye contact. Most older children and adults with AS are able to communicate by pointing and reaching, using gestures, pointing to body parts, and by using communication boards. Effective fluent use of sign language does not occur [Larson et al 2015, Pearson et al 2019]. However, some individuals with mosaic imprinting center defects have considerable language, speaking in short sentences and using up to 60 words [Fairbrother et al 2015, Le Fevre et al 2017].
Feeding and gastrointestinal issues. Young infants with AS may have difficulties with breast feeding or bottle feeding (as a result of sucking difficulties) and hypotonia. Almost 50% demonstrate poor feeding – particularly those with the 15q11.2-q13 deletion. Approximately 10%-15% require a gastrostomy tube or nasogastric tube [Glassman et al 2017, Khan et al 2019, Bindels-de Heus et al 2020].
Gastroesophageal reflux disease (GERD) occurs in 45%-65% of individuals with AS, resulting in poor weight gain and emesis in infants [Glassman et al 2017, Khan et al 2019]. Parents may first notice difficulty swallowing or spitting up, trouble breathing, gagging, back arching, refusal to feed, pain, and discomfort with feeding. Treatment is necessary to prevent upper gastrointestinal bleeding and esophagitis. Issues related to GERD can continue to be a problem throughout life [Larson et al 2015].
Vomiting is not uncommon and can be either cyclic or intermittent. Cyclic vomiting appears to be more common in individuals with a 15q11.2-q13 deletion or uniparental disomy (UPD). Vomiting (unrelated to illness or food allergies) can be due to a variety of factors including anxiety and behavior issues, side effects of medication, and constipation [Glassman et al 2017].
Hyperphagia and problematic food-related behaviors are seen in all genetic subtypes with a prevalence of 20%-50% [Welham et al 2015, Bindels-de Heus et al 2020].
Constipation is common and can occur at any age. Symptoms include hard or infrequent stools, poor or worsening appetite, vomiting, and stomach pain. Appropriate and timely management is important, as constipation can result in behavioral changes, weight loss, poor sleep quality, and increased seizures [Glassman et al 2017, Khan et al 2019].
Microcephaly. Delayed or disproportionately slow head growth usually results in absolute or relative microcephaly (< -2 SD) by age two years, often accompanied by a flattened occiput. The reported frequency of microcephaly varies from 25% to 80%. Microcephaly is more prevalent in individuals with the 15q11.2-q13 deletion. A normal head circumference should not exclude Angelman syndrome as a possible diagnosis [Tan et al 2011, Bindels-de Heus et al 2020].
Characteristic facial features. Flat occiput, occipital groove, wide mouth, widely spaced teeth, protruding tongue, and prognathia are reported in fewer than 80% of affected individuals (see Figure 1). Individuals with AS can have lighter hair, skin, and eyes relative to family members, particularly those with the 15q11.2-q13 deletion.
Although the tongue is normal in shape and size, about 30%-50% have persistent tongue protrusion. For some individuals, the problem persists into adulthood. Drooling can lead to skin irritation and aspiration, but is generally not associated with significant complications. Surgical or medication treatments (e.g., surgical reimplantation of the salivary ducts or use of local scopolamine patches) are generally not effective. Treatment is usually conservative including bibs and sometimes occupational therapy [Boyce & Bakheet 2005, Scully et al 2009].
Strabismus and other eye findings. The incidence of strabismus is 40%-50%, regardless of molecular cause [Tan et al 2011, Khan et al 2019, Bindels-de Heus et al 2020]. OCA2 is located in the 15q11.2-q13 region and has a role in pigmentation of the skin, hair, and irides. Strabismus appears to be more common in genetic disorders that cause ocular hypopigmentation. Pigment in the retina is crucial for normal development of the optic nerve pathways. Although ocular hypopigmentation is reported in the iris and choroid (not the fovea) in individuals with AS, hypopigmentation can be found in individuals without the 15q11.2-q13 deletion and OCA2 may not be the sole explanation. Standard treatments are used for strabismus including glasses, patching, and surgery when appropriate. Hypermotoric activities can make compliance challenging. Approximately 30% require strabismus surgery with overall successful outcomes [Ye et al 2019].
Astigmatism is the most common refractive error. Keratoconus can occur and may be secondary to persistent eye rubbing or gouging behaviors or other causes. Additional ocular findings include myopia, hyperopia, nystagmus, optic nerve atrophy or optic disk pallor, retinochoroidal atrophy, ptosis, and amblyopia [Michieletto et al 2011].
Orthopedics. Scoliosis can develop in adolescence and becomes more common with advancing age [Giroud et al 2015, Larson et al 2015]. Approximately 10%-20% of children develop scoliosis. At least 30%-50% of adults have scoliosis that is typically thoracic. Increased lumbar lordosis is reported in 20%-25% of adults [Sachdeva et al 2016, Prasad et al 2018]. Scoliosis can limit mobility and is treated with bracing to prevent progression. Surgical correction may be necessary for individuals with severe scoliosis. Additional orthopedic complications include hip dysplasia and low bone mineral density. Osteopenia appears to be more common in those treated with anti-seizure medication [Coppola et al 2007, Larson et al 2015].
Pubertal development is generally normal in individuals with AS. Larson et al [2015] reported early menarche in 17% and late menarche in 27%. Hormonal changes during puberty can affect both behavior and epilepsy. Fertility appears to be normal and procreation appears possible for both males and females. Discussion with a gynecologist is appropriate to explore options to regulate menses [Kaskowitz et al 2016]. Lossie & Driscoll [1999] reported transmission of a 15q11.2-q13 deletion to a fetus by a mother with AS.
Growth. Length, weight, and head circumference at birth are usually normal. Average height during childhood in individuals with AS is lower than in the general population, but most have a normal final adult height. Individuals with AS have an increased weight-to-height ratio, which is more frequently reported in individuals without the 15q11.2-q13 deletion [Mertz et al 2014, Carson et al 2019, Bindels-de Heus et al 2020].
Prognosis. Young adults appear to have generally good physical health. Seizures, abnormal movements (ataxia, decreased ambulation), and difficult behaviors may continue throughout adulthood, as well as other described gastrointestinal, sleep, and orthopedic issues. With respect to other health issues, adults with AS appear to be at the same risk as the general population. Independent living is not possible for adults with AS. Many live at home or in home-like placements. Life span data are not available, but life span appears to be nearly normal [Larson et al 2015].
Genotype-Phenotype Correlations
All molecular causes of AS lead to a similar phenotype of severe-to-profound intellectual disability, movement disorder, characteristic behaviors, and severe limitations in speech and language. However, some phenotypic differences correlate with genotype [Tan et al 2011, Valente et al 2013, Bindels-de Heus et al 2020, Keute et al 2020]. These correlations are broadly summarized here:
- The 5- to 7-Mb 15q11.2-q13 deletion results in the most severe phenotype with microcephaly, seizures, motor difficulties (e.g., ataxia, hypotonia, feeding difficulties), and language impairment. These individuals also have lower body mass index compared to individuals with UPD or an imprinting defect. It is unclear if individuals with larger deletions (e.g., BP1-BP3 [class I; ISCA-37404] break points) can be clinically distinguished from those with BP2-BP3 (class II; ISCA-37478) break points (see Figure 2).
- Individuals with UBE3A pathogenic variants and those with imprinting defects may be less clinically affected that those with UPD.
- UBE3A truncating variants may cause more severe clinical manifestations than UBE3A missense variants [Keute et al 2020].
- Individuals who are mosaic for nondeletion imprinting defects (~20% of those with an imprinting defect) have the most advanced speech abilities [Nazlican et al 2004]; they may speak up to 50-60 words and use simple sentences [Fairbrother et al 2015, Le Fevre et al 2017].
- Individuals with a 15q11.2-q13 deletion including OCA2 frequently have hypopigmented irides, skin, and hair. OCA2 encodes a protein important in tyrosine metabolism that is associated with the development of pigment in the skin, hair, and irides (see Oculocutaneous Albinism Type 2). However, other factors in addition to OCA2 haploinsufficiency appear to account for the relative hypopigmentation in individuals with AS, as UBE3A has been shown to modulate melanocortin 1 receptor (MC1R) activity in somatic tissues [Low & Chen 2011].

Penetrance
UBE3A pathogenic variants, imprinting center deletions, very small 15q11.2-q13 deletions that include UBE3A [Kuroda et al 2014], and certain chromosome translocations affecting the paternal allele may be non-penetrant (see Figure 3).
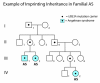
Prevalence
The population prevalence of AS is estimated at 1:12,000-1:24,000 [Mertz et al 2013].
Genetically Related (Allelic) Disorders
Prader-Willi syndrome (PWS) is caused by an absence of expression of imprinted genes in the paternally contributed 15q11.2-q13 region. Although PWS and Angelman syndrome (AS) are clinically distinct in older children, some clinical overlap exists (e.g., feeding difficulties, hypotonia, developmental delay) in children younger than age two years.
Interstitial duplications of 15q11.2-q13 on the maternally derived chromosome cause a disorder clinically distinct from AS and PWS. Individuals with dup15q11.2-q13 do not have facial dysmorphism but have mild to moderately severe learning deficits and may have behaviors in the autism spectrum. See 15q Duplication Syndrome and Related Disorders.
Differential Diagnosis
Infants with Angelman syndrome (AS) commonly present with nonspecific psychomotor delay and/or seizures; therefore, the differential diagnosis is broad and nonspecific, encompassing such entities as cerebral palsy, static encephalopathy, or mitochondrial encephalomyopathy. The tremulousness and jerky limb movements seen in most infants with AS may help distinguish AS from these conditions.
AS-mimicking conditions have been reviewed [Tan et al 2014] and many of these, as well as additional disorders to consider in the differential diagnosis, are listed in Table 3.
Table 3.
Genes of Interest in the Differential Diagnosis of Angelman Syndrome
Some chromosome disorders may mimic features of AS including Phelan-McDermid syndrome (22q13.3 deletion), Koolen-de Vries syndrome (associated with either a 17q21.31 deletion or a heterozygous intragenic KANSL1 pathogenic variant), 1q21.1 recurrent microdeletion, and others. These disorders are characterized by nondysmorphic facial features, language and intellectual impairment, and (in some individuals) happy or excitable behaviors.
Note: Infants with AS who present with feeding difficulties and hypotonia may be misdiagnosed as having Prader-Willi syndrome if a 15q11.2-q13 deletion, detected by chromosomal microarray or FISH, was not proven by DNA methylation analysis to be of maternal origin (see Genetically Related Disorders).
Management
No clinical practice guidelines for Angelman syndrome (AS) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with AS, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Angelman Syndrome
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with Angelman Syndrome
Surveillance
Table 6.
Recommended Surveillance for Individuals with Angelman Syndrome
Agents/Circumstances to Avoid
Overtreatment
- Children with AS are at risk for medication overtreatment because their movement abnormalities can be mistaken for seizures and because EEG abnormalities can persist even when seizures are controlled.
- The behavioral phenotype of AS includes hyperexcitability, hypermotoric behaviors, and deficits in social communication. These limitations place affected individuals at risk for social disruptions. On occasion, the use of risperidone (Risperdal®) or other atypical antipsychotic drugs provides some but often limited benefit. When such drugs are needed, care must be taken to avoid oversedation and other side effects.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Clinical trials involving oral administration of folate, vitamin B12, creatine, and betaine have been undertaken in an attempt to augment DNA methylation pathways and possibly increase expression of the paternal UBE3A allele in the central nervous system; however, the initial trial did not demonstrate significant clinical benefit [Peters et al 2010]. An ongoing study using oral gaboxadol, a highly selective GABA receptor agonist, has not yet shown clear benefit [Bird et al 2021]. A study of oral levodopa/carbidopa did not show significant benefit in AS [Tan et al 2018]. Two clinical trials are currently evaluating antisense oligonucleotide-mediated enhancement of UBE3A expression: one trial is sponsored by Hoffmann‐La Roche (molecule RO7248824; NCT04428281) and the other is sponsored by GeneTX Biotherapeutics (molecule GTX-102; NCT04259281).
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Individuals with Angelman syndrome (AS) typically represent simplex cases (i.e., a single affected family member) and have the disorder as the result of a de novo genetic alteration associated with a very low recurrence risk (e.g., deletion of the maternally inherited 15q11.2-q13 region). Less commonly, an individual with AS has the disorder as the result of a genetic alteration associated with an imprinting pattern of autosomal dominant inheritance (e.g., a pathogenic variant in UBE3A) or variable recurrence risk (e.g., an unbalanced chromosome translocation). Reliable recurrence risk assessment therefore requires identification of the underlying genetic mechanism in the proband and confirmation of the genetic status of the parents.
Risk to Family Members
Parents of a proband. The parents of a proband are unaffected.
Sibs of a proband. The risk to the sibs of an individual with AS depends on the genetic mechanism of AS in the proband and the genetic status of the parents:
- If the proband does not have a UBE3A pathogenic variant and the DNA methylation pattern is characteristic for absence of the maternal contribution, the underlying genetic mechanism (i.e., deletion of the maternally inherited 15q11.2-q13 region, uniparental disomy [UPD] of the paternal chromosome region 15q11.2-q13, or an imprinting defect of the maternal chromosome 15q11.2-q13 region) should be determined for genetic counseling purposes; recommended testing for the proband is as follows:
- 15q11.2-q13 deletion analysis (by chromosomal microarray or other methods) should be performed first.
- If a 15q11.2-q13 deletion is not detected and the microarray (or other method used to detect copy number variants) does not identify segmental or whole-chromosome isodisomy, analysis of DNA polymorphisms on chromosome 15 can be used to rule out a whole-chromosome heterodisomy (to the authors' knowledge, this type of heterodisomy has not been reported in AS, although it is relatively common in Prader-Willi syndrome [Fridman & Koiffmann 2000]).
- If UPD is not detected, the presumption is that an imprinting defect is present; additional studies can then determine if there is a deletion in the imprinting center.
- Once the underlying genetic mechanism has been established in the proband, the genetic status of the parents can be assessed.
- Recommendations for parental testing (based on the genetic mechanism in the proband) and corresponding recurrence risks to sibs are summarized in Table 7.
Table 7.
Risks to Sibs of a Proband with Angelman Syndrome by Genetic Mechanism and Parental Genetic Status
Offspring of a proband. To date, only one individual with AS has been reported to have reproduced [Lossie & Driscoll 1999]. The risk to offspring should be determined in the context of formal genetic counseling.
Other family members
- If a UBE3A pathogenic variant, imprinting center deletion, or structural chromosome rearrangement has been identified in the mother (or father in the case of UPD and Robertsonian translocations) of a proband, the sibs of the parent with the predisposing genetic alteration should be offered genetic counseling and the option of genetic testing.
- If a proband's mother is heterozygous for a known imprinting center deletion or UBE3A pathogenic variant, the mother's sibs are also at risk of having the imprinting center deletion or the UBE3A pathogenic variant. Each child of the unaffected heterozygous sister is at a 50% risk of having AS. Unaffected maternal uncles of the proband who are heterozygous are not at risk of having affected children, but are at risk of having affected grandchildren through their unaffected daughters who inherited the imprinting center deletion or UBE3A pathogenic variant from them.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are at risk of having children with AS.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown).
Prenatal Testing and Preimplantation Genetic Testing
Prenatal testing and preimplantation genetic testing for high-risk pregnancies require prior identification of the underlying genetic mechanism in the proband.
High risk. Prenatal detection of all the known molecular genetic alterations (i.e., molecular classes Ia, Ib, IIa, IIb, IIIa, IIIb, IV; see Table 7) in the 15q11.2-q13 region that give rise to AS is possible through DNA and/or chromosome/FISH analysis of fetal cells obtained by chorionic villus sampling (CVS) or amniocentesis.
DNA methylation analysis (for 5- to 7-Mb deletion of 15q11.2-q13, UPD, and imprinting center defects) on fetal cells obtained by CVS is theoretically possible. However, the few clinical laboratories doing prenatal testing using DNA methylation analysis prefer to use amniocytes because of the relative hypomethylation of cells derived from the placenta. FISH analysis, imprinting center deletion analysis, and sequence analysis of UBE3A should be technically possible using fetal cells obtained by CVS [Beygo et al 2019].
Prenatal testing should be undertaken only after the genetic mechanism in the index case has been established and the couple has been counseled regarding recurrence risk, as the risks and the type of molecular genetic testing used vary according to the type of molecular defect in the proband (see Establishing the Diagnosis).
- Parents with normal chromosomes who have had one child with AS caused by either 15q11.2-q13 deletion or UPD have a low recurrence risk but may be offered prenatal testing for reassurance.
- Parents who have had one child with AS caused by a UBE3A pathogenic variant should be offered prenatal testing even if the mother does not have a UBE3A pathogenic variant because of the possibility of maternal germline mosaicism.
- Prenatal testing for an inherited translocation involving chromosome 15 is relevant because of the increased recurrence risk. FISH analysis and parent-of-origin (DNA methylation and/or polymorphism) studies should be considered if an inherited translocation involving chromosome 15 is present.
Low risk. For low-risk pregnancies with no family history of AS, AS needs to be considered in the following instances:
- If a 15q11.2-q13 deletion is suspected on cytogenetic studies from CVS or amniocentesis, FISH analysis or chromosomal microarray analysis (CMA) is indicated to confirm the deletion. If the 15q11.2-q13 deletion is confirmed, parent-of-origin studies [Beygo et al 2019] can be performed to determine if the 15q11.2-q13 deletion is maternally derived (fetus has AS) or paternally derived (fetus has Prader-Willi syndrome [PWS]).
- If trisomy 15 or mosaic trisomy 15 is detected on CVS, and if subsequent amniocentesis reveals 46 chromosomes, the possibility of trisomy rescue leading to AS (paternal UPD) or PWS (maternal UPD) through the loss of a parental chromosome 15 must be considered. In this instance, parent-of-origin (DNA) studies on amniocytes can be performed.
- If a de novo translocation involving chromosome 15 or a supernumerary chromosome 15 marker is detected, FISH analysis or CMA and parent-of-origin studies should be considered to evaluate for a possible 15q11.2-q13 deletion (of variable size) or UPD.
Preimplantation genetic testing (PGT) may be an option for families in which the underlying mechanism has been identified in the proband to be a UBE3A pathogenic variant or an imprinting center deletion. (The relative hypomethylation of the early embryo makes PGT problematic for DNA methylation testing.)
Other
Assisted reproductive technology (ART). In vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI) have been demonstrated to increase the chance of certain imprinting disorders (e.g., Beckwith-Wiedemann syndrome) in offspring. A 2018 report detected one occurrence of AS caused by an imprinting error in 949 pregnancies analyzed for AS following IVF. Although the researchers hypothesized that there may be an increased risk in IVF of imprinting errors, the study was limited by its small sample size and number of participating prenatal centers [Johnson et al 2018]. Additional studies have demonstrated no significant association between AS and IVF or ICSI [Vermeiden & Bernardus 2013, Hattori et al 2019].
Fertility. Research from the Netherlands and Germany demonstrates an association between fertility issues and the incidence of AS. The percent of couples who experienced fertility issues before having a child with AS ranged from 19% to 25%. A positive association with fertility issues and AS was not identified in families queried in the United Kingdom [Vermeiden & Bernardus 2013].
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Angelman Syndrome Foundation, Inc. (ASF)4255 Westbrook DriveSuite 219Aurora IL 60504Phone: 800-432-6435 (toll-free); 630-978-4245Fax: 630-978-7408Email: info@angelman.org
- Foundation for Angelman Syndrome Therapeutics (FAST)PO Box 608Downers Grove IL 60515Phone: 630-852-FAST; 866-783-0078Fax: 630-852-3270Email: info@CureAngelman.org
- Medical Home Portal
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- American Epilepsy Society
- Epilepsy FoundationPhone: 301-459-3700Fax: 301-577-2684
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Angelman Syndrome: Genes and Databases
Table B.
OMIM Entries for Angelman Syndrome (View All in OMIM)
Molecular Pathogenesis
The cardinal features of Angelman syndrome result from deficient expression or function of the maternally inherited UBE3A allele [Jiang et al 1999, Lossie et al 2001, Nicholls & Knepper 2001]. UBE3A encodes ubiquitin-protein ligase E3A, a protein involved in the ubiquitination pathway, which targets selected proteins for degradation. UBE3A displays predominant maternal expression in human fetal brain and adult frontal cortex [Rougeulle et al 1997, Vu & Hoffman 1997, Herzing et al 2001].
UBE3A has a large 5' CpG island; DNA methylation does not differ between the maternal and paternal alleles. Because no differentially methylated region is present in UBE3A, imprinted expression of UBE3A is regulated indirectly through a paternally expressed antisense transcript. Runte et al [2001] have shown that a long SNURF-SNRPN sense/UBE3A antisense RNA transcript exists in the AS/PWS region, starting from the SNURF-SNRPN imprinting center and extending more than 460 kb to at least the 5' end of UBE3A. This antisense transcript can block paternal UBE3A expression [Meng et al 2013].
Disruption of UBE3A could affect crucial neuronal processes of protein degradation, replacement, and/or regulation that would otherwise be balanced or maintained by a functional ubiquitin-proteasome system. The ubiquitin-proteasome pathway is essential for cellular functioning including signal transduction, cell-cycle progression, DNA repair, and transcriptional regulation [Ciechanover 1998, Hershko & Ciechanover 1998].
Pathogenic variants
- Deletions of 15q11.2-q13 (65%-75%). Three chromosome break points characterized by low copy repeat regions (proximal BP1, BP2, and a distal BP3) are involved in most AS-causing deletion events involving 15q11.1-q13. These deletions span approximately 5-7 Mb [Amos-Landgraf et al 1999, Christian et al 1999] (see Figure 2). Fewer than 10% of individuals with 15q11.2-q13 deletions have a deletion extending from the BP1/BP2 region to more distal low copy repeat regions, BP4 or BP5 (see Figure 2) [Sahoo et al 2007].Note: Microdeletions that flank the typical deletion region and include areas between BP1 and BP2 [Doornbos et al 2009], BP3 and BP4 [Rosenfeld et al 2011], and the more distal microdeletion syndrome involving region 15q13.3 [Masurel-Paulet et al 2010] have been described. However, individuals with these deletions do not exhibit features of AS.
- Genomic abnormalities of 15q11.1-q13. It is possible that in otherwise healthy individuals, preexisting genomic abnormalities may predispose to deletion of 15q11.1-q13 in the germline, resulting in offspring with AS.
- A proportion of mothers who have a child with AS due to a 15q11.1-q13 deletion have been found to have inversions in the 15q11.2-q13 region [Gimelli et al 2003].
- A kindred in which two individuals had deletions (one deletion causing PWS and the other causing AS) has been previously reported to be associated with an inherited inverted intrachromosomal insertion of 15q11.2-q13 [Collinson et al 2004].
- Paternal uniparental disomy of chromosome 15 (3%-7%). In contrast to PWS, the paternal UPD observed in AS is most likely to be postzygotic in origin [Fridman & Koiffmann 2000, Robinson et al 2000]. Paternal UPD of meiotic origin does occur but this mechanism is less common than the maternal UPD associated with PWS.
- Imprinting defects (3%). This subset of individuals with AS have an imprinting defect that disrupts the resetting of the normal imprint during gametogenesis. Even though these individuals have biparental inheritance of chromosome 15, the maternal 15q11.2-q13 region has a paternal epigenotype and is, therefore, transcriptionally incompetent for the maternal-only expressed gene(s) in this region [Buiting et al 2016].Mapping these imprinting center deletions (as well as mapping the imprinting center deletions that are associated with PWS) has delineated two small regions of deletion overlap (SRO) that define two critical elements in the imprinting center, the AS-SRO and the PWS-SRO [Buiting et al 1995] (see Figure 2). The PWS-SRO is 4.3 kb and overlaps with the SNURF-SNRPN exon1/promoter region [Ohta et al 1999]. Imprinting center deletions in individuals with AS affect the more centromeric SNURF-SNRPN promoter/exon 1 region. The AS-SRO is 880 bp and is 35 kb proximal to SNURF-SNRPN exon 1 [Buiting et al 2016]. Most individuals with AS caused by imprinting defects do not have a deletion of the AS imprinting center, but rather have epigenetic defects that disrupt imprinting center function.
UBE3A (~11%). More than 250 pathogenic variants have been reported including small deletions and duplications leading to frameshifts, missense and nonsense pathogenic variants, splicing defects, large deletions, and complex rearrangements.
Chapter Notes
Author History
Adati I Dagli, MD (2008-present)
Hui-Ja Dong; University of Florida College of Medicine (2003-2005)
Daniel J Driscoll, PhD, MD; University of Florida College of Medicine (1998-2011)
Amy C Lossie, PhD; University of Florida College of Medicine (1998-2003)
Jennifer Mathews, MS, CGC (2015-present)
Charles A Williams, MD (1998-present)
Revision History
- 22 April 2021 (sw) Comprehensive update posted live
- 21 December 2017 (aa) Revision: WAC-related intellectual disability added to Differential Diagnosis
- 14 May 2015 (me) Comprehensive update posted live
- 16 June 2011 (me) Comprehensive update posted live
- 5 September 2008 (me) Comprehensive update posted live
- 30 July 2007 (cd) Revision: targeted mutation analysis no longer available clinically
- 21 February 2007 (cd) Revision: clarification of Genetic Counseling section
- 8 November 2005 (me) Comprehensive update posted live
- 3 September 2004 (cw) Author revisions
- 29 July 2003 (me) Comprehensive update posted live
- 2 April 2002 (cw) Author revision
- 21 November 2000 (me) Comprehensive update posted live
- 15 September 1998 (pb) Review posted live
- April 1998 (cw) Original submission
References
Published Guidelines / Consensus Statements
- Beygo J, Buiting K, Ramsden SC, Ellis R, Clayton-Smith J, Kanber D. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Available online. 2019. Accessed 7-5-22. [PMC free article: PMC6777528] [PubMed: 31235867]
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413–18. [PubMed: 16470747]
Literature Cited
- Amos-Landgraf JM, Ji Y, Gottlieb W, Depinet T, Wandstrat AE, Cassidy SB, Driscoll DJ, Rogan PK, Schwartz S, Nicholls RD. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet. 1999;65:370–86. [PMC free article: PMC1377936] [PubMed: 10417280]
- Arn PH, Williams CA, Zori RT, Driscoll DJ, Rosenblatt DS. Methylenetetrahydrofolate reductase deficiency in a patient with phenotypic findings of Angelman syndrome. Am J Med Genet. 1998;77:198–200. [PubMed: 9605586]
- Arron K, Oliver C, Moss J, Berg K, Burbidge C. The prevalence and phenomenology of self-injurious and aggressive behaviour in genetic syndromes. J Intellect Disabil Res. 2011;55:109–20. [PubMed: 20977515]
- Beygo J, Buiting K, Ramsden SC, Ellis R, Clayton-Smith J, Kanber D. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur J Hum Genet. 2019;27:1326–40. [PMC free article: PMC6777528] [PubMed: 31235867]
- Bindels-de Heus K, Mous SE, Ten Hooven-Radstaake M, van Iperen-Kolk BM, Navis C, Rietman AB, Ten Hoopen LW, Brooks AS, Cf AS EE, Elgersma Y, Moll HA, de Wit MY. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. 2020;182:53–63. [PMC free article: PMC6916553] [PubMed: 31729827]
- Bird LM. Angelman syndrome: review of clinical and molecular aspects. The application of clinical genetics. 2014;7:93–104. [PMC free article: PMC4036146] [PubMed: 24876791]
- Bird LM, Ochoa-Lubinoff C, Tan WH, Heimer G, Melmed RD, Rakhit A, Visootsak J, During MJ, Holcroft C, Burdine RD, Kolevzon A, Thibert RL. The STARS phase 2 study: a randomized controlled trial of gaboxadol in Angelman syndrome. Neurology. 2021;96:e1024–e1035. [PMC free article: PMC8055330] [PubMed: 33443117]
- Boyce HW, Bakheet MR. Sialorrhea: a review of a vexing, often unrecognized sign of oropharyngeal and esophageal disease. J Clin Gastroenterol. 2005;39:89–97. [PubMed: 15681902]
- Boyd SG, Harden A, Patton MA. The EEG in early diagnosis of the Angelman (happy puppet) syndrome. Eur J Pediatr. 1988;147:508–13. [PubMed: 3409926]
- Bramswig NC, Buiting K, Bechtel N, Horsthemke B, Rostasy K, Wieczorek D. Angelman syndrome-affected individual with a numerically normal karyotype and isodisomic paternal uniparental disomy of chromosome 15 due to maternal Robertsonian translocation (14;15) by monosomy rescue. Cytogenet Genome Res. 2018;156:9–13. [PubMed: 30016768]
- Buiting K, Barnicoat A, Lich C, Pembrey M, Malcolm S, Horsthemke B. Disruption of the bipartite imprinting center in a family with Angelman syndrome. Am J Hum Genet. 2001;68:1290–4. [PMC free article: PMC1226110] [PubMed: 11283796]
- Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, Horsthemke B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9:395–400. [PubMed: 7795645]
- Buiting K, Williams C, Horsthemke B. Angelman syndrome - insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016;12:584–93. [PubMed: 27615419]
- Buntinx IM, Hennekam RC, Brouwer OF, Stroink H, Beuten J, Mangelschots K, Fryns JP. Clinical profile of Angelman syndrome at different ages. Am J Med Genet. 1995;56:176–83. [PubMed: 7625442]
- Carson RP, Bird L, Childers AK, Wheeler F, Duis J. Preserved expressive language as a phenotypic determinant of Mosaic Angelman Syndrome. Mol Genet Genomic Med. 2019;7:e837. [PMC free article: PMC6732290] [PubMed: 31400086]
- Castro-Gago M, Gomez-Lado C, Eiris-Punal J, Rodriguez-Mugico VM. Abnormal myelination in Angelman syndrome. Eur J Paediatr Neurol. 2010;14:292. [PubMed: 19720548]
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet. 1999;8:1025–37. [PubMed: 10332034]
- Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–60. [PMC free article: PMC1171061] [PubMed: 9857172]
- Clayton-Smith J. Clinical research on Angelman syndrome in the United Kingdom: observations on 82 affected individuals. Am J Med Genet. 1993;46:12–15. [PubMed: 7684188]
- Collinson MN, Roberts SE, Crolla JA, Dennis NR. A familial balanced inverted insertion ins(15)(q15q13q11.2) producing Prader-Willi syndrome, Angelman syndrome and duplication of 15q11.2-q13 in a single family: Importance of differentiation from a paracentric inversion. Am J Med Genet A. 2004;126A:27–32. [PubMed: 15039970]
- Coppola G, Verrotti A, Mainolfi C, Auricchio G, Fortunato D, Operto FF, Pascotto A. Bone mineral density in angelman syndrome. Pediatr Neurol. 2007;37:411–16. [PubMed: 18021922]
- Doornbos M, Sikkema-Raddatz B, Ruijvenkamp CA, Dijkhuizen T, Bijlsma EK, Gijsbers AC, Hilhorst-Hofstee Y, Hordijk R, Verbruggen KT, Kerstjens-Frederikse WS, van Essen T, Kok K, van Silfhout AT, Breuning M, van Ravenswaaij-Arts CM. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur J Med Genet. 2009;52:108–15. [PubMed: 19328872]
- Dosier LBM, Vaughn BV, Fan Z. Sleep disorders in childhood neurogenetic disorders. Children (Basel). 2017:4. [PMC free article: PMC5615272] [PubMed: 28895939]
- Elia M. Myoclonic status in nonprogressive encephalopathies: an update. Epilepsia. 2009;50:41–4. [PubMed: 19469845]
- Fairbrother LC, Cytrynbaum C, Boutis P, Buiting K, Weksberg R, Williams C. Mild Angelman syndrome phenotype due to a mosaic methylation imprinting defect. Am J Med Genet A. 2015;167:1565–9. [PubMed: 25899869]
- Fedak Romanowski EM, McNamara NA, Neil EE, Gottlieb-Smith R, Dang LT. Seizure rescue medications for out-of-hospital use in children. J Pediatr. 2021;229:19–25. [PubMed: 33228949]
- Fiumara A, Pittala A, Cocuzza M, Sorge G. Epilepsy in patients with Angelman syndrome. Ital J Pediatr. 2010;36:31. [PMC free article: PMC2865483] [PubMed: 20398390]
- Fridman C, Koiffmann CP. Origin of uniparental disomy 15 in patients with Prader-Willi or Angelman syndrome. Am J Med Genet. 2000;94:249–53. [PubMed: 10995513]
- Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol L, Schinzel A, Estivill X, Zuffardi O. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet. 2003;12:849–58. [PubMed: 12668608]
- Giroud M, Daubail B, Khayat N, Chouchane M, Berger E, Muzard E, Medeiros de Bustos E, Thauvin-Robinet C, Faivre L, Masurel A, Darmency-Stamboul V, Huet F, Bejot Y, Giroud M, Moulin T. Angelman syndrome: a case series assessing neurological issues in adulthood. European neurology. 2015;73:119–25. [PubMed: 25472600]
- Glassman LW, Grocott OR, Kunz PA, Larson AM, Zella G, Ganguli K, Thibert RL. Prevalence of gastrointestinal symptoms in Angelman syndrome. Am J Med Genet A. 2017;173:2703–9. [PubMed: 28816003]
- Harpey JP, Heron D, Prudent M, Lesourd S, Henry I, Royer-Legrain G, Munnich A, Bonnefont JP. Recurrent meiotic nondisjunction of maternal chromosome 15 in a sibship. Am J Med Genet. 1998;76:103–4. [PubMed: 9508076]
- Harting I, Seitz A, Rating D, Sartor K, Zschocke J, Janssen B, Ebinger F, Wolf NI. Abnormal myelination in Angelman syndrome. Eur J Paediatr Neurol. 2009;13:271–6. [PubMed: 18573670]
- Hattori H, Hiura H, Kitamura A, Miyauchi N, Kobayashi N, Takahashi S, Okae H, Kyono K, Kagami M, Ogata T, Arima T. Association of four imprinting disorders and ART. Clin Epigenetics. 2019;11:21. [PMC free article: PMC6367766] [PubMed: 30732658]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. [PubMed: 9759494]
- Herzing LB, Kim SJ, Cook EH Jr, Ledbetter DH. The human aminophospholipid-transporting ATPase gene ATP10C maps adjacent to UBE3A and exhibits similar imprinted expression. Am J Hum Genet. 2001;68:1501–5. [PMC free article: PMC1226137] [PubMed: 11353404]
- Hosoki K, Takano K, Sudo A, Tanaka S, Saitoh S. Germline mosaicism of a novel UBE3A mutation in Angelman syndrome. Am J Med Genet A. 2005;138A:187–9. [PubMed: 16100729]
- Jiang Y, Lev-Lehman E, Bressler J, Tsai TF, Beaudet AL. Genetics of Angelman syndrome. Am J Hum Genet. 1999;65:1–6. [PMC free article: PMC1378067] [PubMed: 10364509]
- Johnson JP, Schoof J, Beischel L, Schwancke C, Goldberg J, Black L, Ross L, Bhatt S. Detection of a case of Angelman syndrome caused by an imprinting error in 949 pregnancies analyzed for AS following IVF. J Assist Reprod Genet. 2018;35:981–4. [PMC free article: PMC6030013] [PubMed: 29654525]
- Kaskowitz AP, Dendrinos M, Murray PJ, Quint EH, Ernst S. The effect of menstrual issues on young women with Angelman syndrome. J Pediatr Adolesc Gynecol. 2016;29:348–52. [PMC free article: PMC4915967] [PubMed: 26718530]
- Kawano O, Egawa K, Shiraishi H. Perampanel for nonepileptic myoclonus in Angelman syndrome. Brain Dev. 2020;42:389–92. [PubMed: 32164978]
- Keute M, Miller MT, Krishnan ML, Sadhwani A, Chamberlain S, Thibert RL, Tan WH, Bird LM, Hipp JF. Angelman syndrome genotypes manifest varying degrees of clinical severity and developmental impairment. Mol Psychiatry. 2020 Epub ahead of print. [PMC free article: PMC8505254] [PubMed: 32792659]
- Khan N, Cabo R, Tan WH, Tayag R, Bird LM. Healthcare burden among individuals with Angelman syndrome: findings from the Angelman Syndrome Natural History Study. Mol Genet Genomic Med. 2019;7:e00734. [PMC free article: PMC6625091] [PubMed: 31090212]
- Kuroda Y, Ohashi I, Saito T, Nagai J, Ida K, Naruto T, Wada T, Kurosawa K. Deletion of UBE3A in brothers with Angelman syndrome at the breakpoint with an inversion at 15q11.2. Am J Med Genet A. 2014;164A:2873–8. [PubMed: 25099823]
- Larson AM, Shinnick JE, Shaaya EA, Thiele EA, Thibert RL. Angelman syndrome in adulthood. Am J Med Genet A. 2015;167A:331–44. [PMC free article: PMC5534346] [PubMed: 25428759]
- Le Fevre A, Beygo J, Silveira C, Kamien B, Clayton-Smith J, Colley A, Buiting K, Dudding-Byth T. Atypical Angelman syndrome due to a mosaic imprinting defect: case reports and review of the literature. Am J Med Genet A. 2017;173:753–7. [PubMed: 28211971]
- Lossie AC, Driscoll DJ. Transmission of Angelman syndrome by an affected mother. Genetics in Medicine. 1999;1:262–6. [PubMed: 11258627]
- Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, Hutson A, Nicholls RD, Zori RT, Williams CA, Driscoll DJ. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001;38:834–45. [PMC free article: PMC1734773] [PubMed: 11748306]
- Low D, Chen KS. UBE3A regulates MC1R expression: a link to hypopigmentation in Angelman syndrome. Pigment Cell Melanoma Res. 2011;24:944–52. [PubMed: 21733131]
- Masurel-Paulet A, Andrieux J, Callier P, Cuisset JM, Le Caignec C, Holder M, Thauvin-Robinet C, Doray B, Flori E, Alex-Cordier MP, Beri M, Boute O, Delobel B, Dieux A, Vallee L, Jaillard S, Odent S, Isidor B, Beneteau C, Vigneron J, Bilan F, Gilbert-Dussardier B, Dubourg C, Labalme A, Bidon C, Gautier A, Pernes P, Pinoit JM, Huet F, Mugneret F, Aral B, Jonveaux P, Sanlaville D, Faivre L. Delineation of 15q13.3 microdeletions. Clin Genet. 2010;78:149–61. [PubMed: 20236110]
- Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet. 2013;9:e1004039. [PMC free article: PMC3873245] [PubMed: 24385930]
- Mertz LG, Christensen R, Vogel I, Hertz JM, Nielsen KB, Gronskov K, Ostergaard JR. Angelman syndrome in Denmark. birth incidence, genetic findings, and age at diagnosis. Am J Med Genet A. 2013;161A:2197–203. [PubMed: 23913711]
- Mertz LG, Christensen R, Vogel I, Hertz JM, Ostergaard JR. Eating behavior, prenatal and postnatal growth in Angelman syndrome. Research in developmental disabilities. 2014;35:2681–90. [PubMed: 25064682]
- Michieletto P, Bonanni P, Pensiero S. Ophthalmic findings in Angelman syndrome. J AAPOS. 2011;15:158–61. [PubMed: 21596294]
- Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res. 2009;53:852–73. [PubMed: 19708861]
- Nazlican H, Zeschnigk M, Claussen U, Michel S, Boehringer S, Gillessen-Kaesbach G, Buiting K, Horsthemke B. Somatic mosaicism in patients with Angelman syndrome and an imprinting defect. Hum Mol Genet. 2004;13:2547–55. [PubMed: 15385437]
- Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–75. [PubMed: 11701647]
- Nolt DH, Mott JM, Lopez WL. Assessment of anticonvulsant effectiveness and safety in patients with Angelman's syndrome using an Internet questionnaire. Am J Health Syst Pharm. 2003;60:2583–7. [PubMed: 14735775]
- Ohta T, Buiting K, Kokkonen H, McCandless S, Heeger S, Leisti H, Driscoll DJ, Cassidy SB, Horsthemke B, Nicholls RD. Molecular mechanism of angelman syndrome in two large families involves an imprinting mutation. Am J Hum Genet. 1999;64:385–96. [PMC free article: PMC1377749] [PubMed: 9973277]
- Pearson E, Wilde L, Heald M, Royston R, Oliver C. Communication in Angelman syndrome: a scoping review. Dev Med Child Neurol. 2019;61:1266–74. [PubMed: 31074506]
- Pelc K, Cheron G, Boyd SG, Dan B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008;9:434–41. [PubMed: 17765640]
- Peters SU, Bird LM, Kimonis V, Glaze DG, Shinawi LM, Bichell TJ, Barbieri-Welge R, Nespeca M, Anselm I, Waisbren S, Sanborn E, Sun Q, O'Brien WE, Beaudet AL, Bacino CA. Double-blind therapeutic trial in Angelman syndrome using betaine and folic acid. Am J Med Genet A. 2010;152A:1994–2001. [PMC free article: PMC3172130] [PubMed: 20635355]
- Peters SU, Goddard-Finegold J, Beaudet AL, Madduri N, Turcich M, Bacino CA. Cognitive and adaptive behavior profiles of children with Angelman syndrome. Am J Med Genet A. 2004;128A:110–13. [PubMed: 15213998]
- Pollack SF, Grocott OR, Parkin KA, Larson AM, Thibert RL. Myoclonus in Angelman syndrome. Epilepsy Behav. 2018;82:170–4. [PubMed: 29555100]
- Prasad A, Grocott O, Parkin K, Larson A, Thibert RL. Angelman syndrome in adolescence and adulthood: A retrospective chart review of 53 cases. Am J Med Genet A. 2018;176:1327–34. [PubMed: 29696750]
- Robinson WP, Christian SL, Kuchinka BD, Penaherrera MS, Das S, Schuffenhauer S, Malcolm S, Schinzel AA, Hassold TJ, Ledbetter DH. Somatic segregation errors predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clin Genet. 2000;57:349–58. [PubMed: 10852369]
- Rosenfeld JA, Stephens LE, Coppinger J, Ballif BC, Hoo JJ, French BN, Banks VC, Smith WE, Manchester D, Tsai AC, Merrion K, Mendoza-Londono R, Dupuis L, Schultz R, Torchia B, Sahoo T, Bejjani B, Weaver DD, Shaffer LG. Deletions flanked by breakpoints 3 and 4 on 15q13 may contribute to abnormal phenotypes. Eur J Hum Genet. 2011;19:547–54. [PMC free article: PMC3083619] [PubMed: 21248749]
- Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17:14–5. [PubMed: 9288088]
- Runte M, Huttenhofer A, Gross S, Kiefmann M, Horsthemke B, Buiting K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet. 2001;10:2687–700. [PubMed: 11726556]
- Sachdeva R, Donkers SJ, Kim SY. Angelman syndrome: a review highlighting musculoskeletal and anatomical aberrations. Clin Anat. 2016;29:561–7. [PubMed: 26480021]
- Sadhwani A, Willen JM, LaVallee N, Stepanians M, Miller H, Peters SU, Barbieri-Welge RL, Horowitz LT, Noll LM, Hundley RJ, Bird LM, Tan WH. Maladaptive behaviors in individuals with Angelman syndrome. Am J Med Genet A. 2019;179:983–92. [PMC free article: PMC8407596] [PubMed: 30942555]
- Sahoo T, Bacino CA, German JR, Shaw CA, Bird LM, Kimonis V, Anselm I, Waisbren S, Beaudet AL, Peters SU. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. 2007;15:943–9. [PubMed: 17522620]
- Samanta D. Epilepsy in Angelman syndrome: a scoping review. Brain Dev. 2021;43:32–44. [PMC free article: PMC7688500] [PubMed: 32893075]
- Sánchez J, Fernandez R, Madruga M, Bernabeu-Wittel J, Antinolo G, Borrego S. Somatic and germ-line mosaicism of deletion 15q11.2-q13 in a mother of dyzigotic twins with Angelman syndrome. Am J Med Genet A. 2014;164A:370–6. [PubMed: 24311297]
- Scully C, Limeres J, Gleeson M, Tomas I, Diz P. Drooling. J Oral Pathol Med. 2009;38:321–7. [PubMed: 19236564]
- Shaaya EA, Grocott OR, Laing O, Thibert RL. Seizure treatment in Angelman syndrome: a case series from the Angelman Syndrome Clinic at Massachusetts General Hospital. Epilepsy Behav. 2016;60:138–41. [PubMed: 27206232]
- Spruyt K, Braam W, Curfs LM. Sleep in Angelman syndrome: a review of evidence. Sleep Med Rev. 2018;37:69–84. [PubMed: 28784434]
- Summers J. Neurodevelopmental outcomes in children with Angelman syndrome after 1 year of behavioural intervention. Dev Neurorehabil. 2012;15:239–52. [PubMed: 22646082]
- Tan WH, Bacino CA, Skinner SA, Anselm I, Barbieri-Welge R, Bauer-Carlin A, Beaudet AL, Bichell TJ, Gentile JK, Glaze DG, Horowitz LT, Kothare SV, Lee HS, Nespeca MP, Peters SU, Sahoo T, Sarco D, Waisbren SE, Bird LM. Angelman syndrome: mutations influence features in early childhood. Am J Med Genet A. 2011;155A:81–90. [PMC free article: PMC3563320] [PubMed: 21204213]
- Tan WH, Bird LM, Sadhwani A, Barbieri-Welge RL, Skinner SA, Horowitz LT, Bacino CA, Noll LM, Fu C, Hundley RJ, Wink LK, Erickson CA, Barnes GN, Slavotinek A, Jeremy R, Rotenberg A, Kothare SV, Olson HE, Poduri A, Nespeca MP, Chu HC, Willen JM, Haas KF, Weeber EJ, Rufo PA. A randomized controlled trial of levodopa in patients with Angelman syndrome. Am J Med Genet A. 2018;176:1099–107. [PMC free article: PMC5867193] [PubMed: 28944563]
- Tan WH, Bird LM, Thibert RL, Williams CA. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A. 2014;164A:975–92. [PubMed: 24779060]
- Tang HS, Wang DG, Xie XM, Li DZ. Apparent germline mosaicism for a 15q11-q13 deletion causing recurrent Angelman syndrome in a Chinese family. Eur J Obstet Gynecol Reprod Biol. 2019;236:255–7. [PubMed: 30890277]
- Thibert RL, Conant KD, Braun EK, Bruno P, Said RR, Nespeca MP, Thiele EA. Epilepsy in Angelman syndrome: a questionnaire-based assessment of the natural history and current treatment options. Epilepsia. 2009;50:2369–76. [PubMed: 19453717]
- Thibert RL, Pfeifer HH, Larson AM, Raby AR, Reynolds AA, Morgan AK, Thiele EA. Low glycemic index treatment for seizures in Angelman syndrome. Epilepsia. 2012;53:1498–502. [PubMed: 22779920]
- Torisu H, Yamamoto T, Fujiwaki T, Kadota M, Oshimura M, Kurosawa K, Akaboshi S, Oka A. Girl with monosomy 1p36 and Angelman syndrome due to unbalanced der(1) transmission of a maternal translocation t(1;15)(p36.3;q13.1). Am J Med Genet A. 2004;131:94–8. [PubMed: 15384094]
- Valente KD, Varela MC, Koiffmann CP, Andrade JQ, Grossmann R, Kok F, Marques-Dias MJ. Angelman syndrome caused by deletion: a genotype-phenotype correlation determined by breakpoint. Epilepsy Research. 2013;105:234–9. [PubMed: 23352739]
- Vermeiden JP, Bernardus RE. Are imprinting disorders more prevalent after human in vitro fertilization or intracytoplasmic sperm injection? Fertility and Sterility. 2013;99:642–51. [PubMed: 23714438]
- Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17:12–3. [PubMed: 9288087]
- Walz NC, Baranek GT. Sensory processing patterns in persons with Angelman syndrome. Am J Occup Ther. 2006;60:472–9. [PubMed: 16915878]
- Walz NC, Beebe D, Byars K. Sleep in individuals with Angelman syndrome: parent perceptions of patterns and problems. AJMR. 2005;110:243–52. [PubMed: 15941362]
- Welham A, Lau J, Moss J, Cullen J, Higgs S, Warren G, Wilde L, Marr A, Cook F, Oliver C. Are Angelman and Prader-Willi syndromes more similar than we thought? Food-related behavior problems in Angelman, Cornelia de Lange, fragile X, Prader-Willi and 1p36 deletion syndromes. Am J Med Genet A. 2015;167A:572–8. [PubMed: 25691410]
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413–18. [PubMed: 16470747]
- Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12:385–95. [PubMed: 20445456]
- Worden L, Grocott O, Tourjee A, Chan F, Thibert R. Diazepam for outpatient treatment of nonconvulsive status epilepticus in pediatric patients with Angelman syndrome. Epilepsy Behav. 2018;82:74–80. [PubMed: 29597185]
- Ye H, Lan X, Liu Q, Zhang Y, Wang S, Zheng C, Di Y, Qiao T. Ocular findings and strabismus surgery outcomes in Chinese children with Angelman syndrome: three case reports. Medicine (Baltimore). 2019;98:e18077. [PMC free article: PMC6940155] [PubMed: 31860958]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: September 15, 1998; Last Update: April 22, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Dagli AI, Mathews J, Williams CA. Angelman Syndrome. 1998 Sep 15 [Updated 2021 Apr 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.


