Summary
Clinical characteristics.
Pitt-Hopkins syndrome (PTHS) is characterized by significant developmental delays with moderate-to-severe intellectual disability and behavioral differences, characteristic facial features, and episodic hyperventilation and/or breath-holding while awake. Speech is significantly delayed and most individuals are nonverbal with receptive language often stronger than expressive language. Other common findings are autism spectrum disorder symptoms, sleep disturbance, stereotypic hand movements, seizures, constipation, and severe myopia.
Diagnosis/testing.
The diagnosis is suspected on clinical findings and confirmed by identification on molecular genetic testing of a heterozygous pathogenic variant in TCF4 or a deletion of the chromosome region in which TCF4 is located (18q21.2).
Management.
Treatment of manifestations: Developmental services for infants (physical, occupational, and speech therapies); individualized education plan for older children with strong consideration for early training in alternative means of communication; behavioral management strategies; possible treatment of abnormal respiratory pattern. Routine management of seizures, myopia, constipation, scoliosis, and ankle instability.
Surveillance: Ongoing developmental assessments to tailor educational services to individual needs; regular follow up with an ophthalmologist to monitor for high myopia and strabismus; periodic reevaluation with a clinical genetics professional regarding current information and recommendations.
Genetic counseling.
PTHS is caused by haploinsufficiency of TCF4 resulting from either a pathogenic variant in TCF4 or a deletion of the chromosome region in which TCF4 is located (18q21.2). Most affected individuals have been simplex cases (i.e., a single occurrence in a family) resulting from a de novo pathogenic variant or deletion. The risk to sibs of a proband is low, but higher than that of the general population because of the possibility of parental germline mosaicism. Once the PTHS-related genetic alteration has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
Pitt-Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, intellectual disability, behavioral differences, distinctive facial features, and unusual breathing patterns.
Suggestive Findings
PTHS should be suspected in individuals with developmental delay, moderate-to-severe intellectual disability, behavioral differences, characteristic facial features, and episodic hyperventilation and/or breath-holding while awake.
Developmental delay / intellectual disability / behavioral differences
- Motor milestones are delayed; often with hypotonia.
- Speech is severely limited to absent, with a history of regression in verbal abilities in some.
- Intellectual disability is typically moderate to severe.
- Autism spectrum disorder (ASD) symptoms are also common, though social engagement is generally present.
- Other behavioral characteristics suggestive of PTHS include the following:
- Love of music
- Frequent smiling
- Stereotypic hand movements
- Hand flapping
- Spontaneous laughter
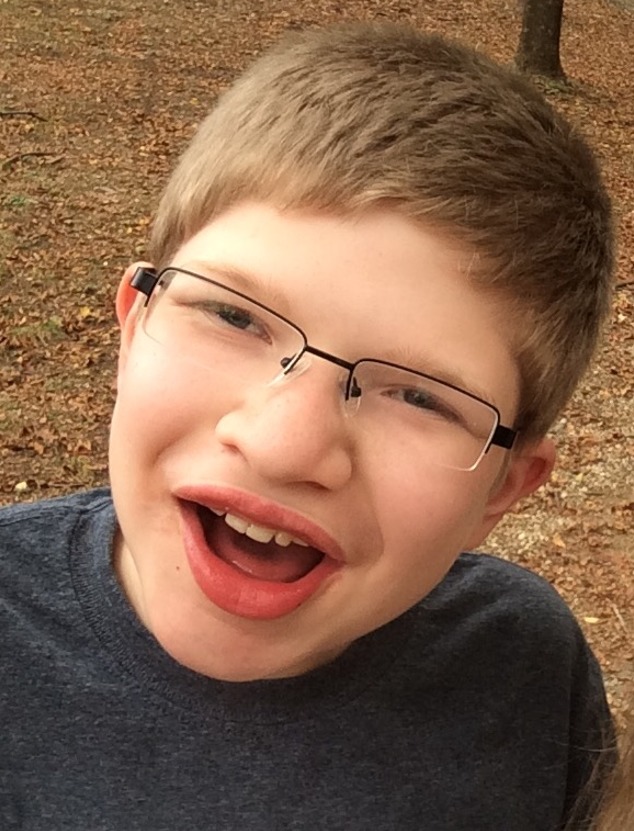
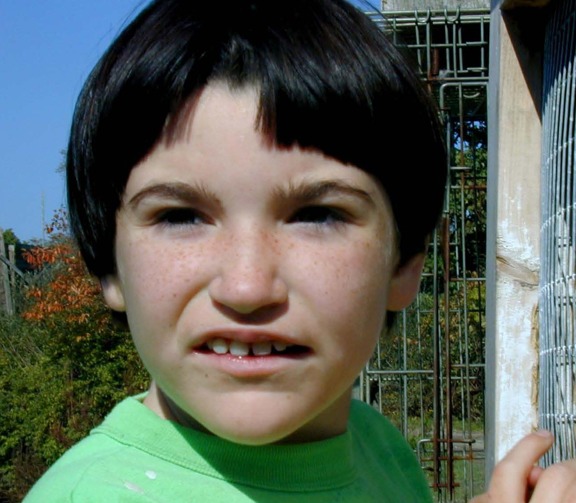
Characteristic facial features that become more apparent with age. The craniofacial features are an important aspect for the diagnosis of PTHS, but may be less obvious in infancy. In many individuals, the prominence of the nose and lower face may be the earliest clue to PTHS in an infant with developmental concerns (see Figures 1, 2, 3, 4, 5).

Figure 1.
Newborn male with Pitt-Hopkins syndrome. Note high nasal root with prominent nasal bridge, depressed nasal tip, and ears with overfolded helix.


Figure 3.
Boy age 13 years with Pitt-Hopkins syndrome (same individual as in Figures 1 and 2). Note wide nasal ridge and alae, short philtrum, and full lips, with everted lower lip.

Figure 4.
Girl age ten years with Pitt-Hopkins syndrome. Note deep-set eyes and depressed nasal tip.

Figure 5.
Woman age 24 years with Pitt-Hopkins syndrome (same individual as in Figure 4). Note prominent nose with depressed tip, short philtrum, full lips, wide mouth with downturned corners, and well-developed chin.
- Deeply set eyes with prominent supraorbital ridges
- Mildly upslanted palpebral fissures
- Broad nasal root, wide nasal ridge, and wide nasal base with enlarged nostrils
- Overhanging or depressed nasal tip, which may be pointed
- Short philtrum
- Thick vermilion of the lower lip, which is often everted
- In some individuals, wide mouth with downturned corners and exaggerated Cupid's bow or tented vermilion of the upper lip
- Widely spaced teeth
- Prominence of the lower face with a well-developed chin. With age, the lower face becomes more prominent and facial features may coarsen.
- Mildly cupped ears with overfolded helices
Episodic hyperventilation and/or breath-holding while awake. Unusual episodes of hyperventilation (which may be followed by apnea) may occur while awake. When present, this finding (in combination with the developmental history and characteristic facial features) is highly suggestive of PTHS; however, the absence of a breathing abnormality should not eliminate consideration of the diagnosis of PTHS as the breathing abnormality may begin sometime in the second half of the first decade, later, or not at all [Zweier et al 2008, Marangi et al 2011].
Note: Information about the presence of the breathing abnormality must be elicited directly: parents may not realize the diagnostic importance of this finding as it would not typically prompt them to seek medical attention [Takano et al 2011; Author, personal observation].
Establishing the Diagnosis
The diagnosis of PTHS is established in a proband with severe intellectual disability and severe speech impairment and confirmed by identification on molecular genetic testing of a heterozygous pathogenic variant in TCF4 or a deletion of the chromosome region in which TCF4 is located (18q21.2) (see Table 1). Rare individuals with a TCF4 pathogenic variant or deletion may lack obvious characteristic facial features, but still manifest intellectual disability and speech impairment.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing or a multigene panel) and genomic testing (comprehensive genomic sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of PTHS is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with intellectual disability / developmental delay are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of PTHS, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of TCF4 is performed first and followed by gene-targeted deletion/duplication analysis if no pathogenic variant is detected on sequence analysis.
- A multigene panel that includes TCF4 and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.For this disorder a multigene panel that includes deletion/duplication analysis as well as sequencing is recommended (see Table 1).
Note: A karyotype should be considered if a diagnosis of PTHS is strongly suspected with normal results on molecular testing, since a chromosome rearrangement disrupting TCF4 without a deletion has been detected in an individual with features of PTHS [Kalscheuer et al 2008, Marangi et al 2011].
Option 2
When the phenotype is indistinguishable from many other inherited disorders with intellectual disability / developmental delay, molecular genetic testing approaches can include comprehensive genomic testing (recommended) and/or gene-targeted testing (multigene panel; to consider):
- Comprehensive genomic testing (when clinically available) includes exome sequencing and genome sequencing.
- A multigene panel for intellectual disability / developmental delay may also be considered.
For each of these options, it would be important to pursue a chromosome microarray (CMA) or a multigene panel that also includes deletion/duplication analysis of TCF4 given the frequency of deletions in this disorder.
Table 1.
Molecular Genetic Testing Used in Pitt-Hopkins Syndrome
Clinical Characteristics
Clinical Description
Children with Pitt-Hopkins syndrome (PTHS) typically present in the first year of life with hypotonia and developmental delays. Some infants have been described as being quiet and "unusually good" with excessive sleeping [Giurgea et al 2008].
Developmental delay / intellectual disability. Developmental delays are significant and intellectual disability is moderate to severe. Hypotonia can be significant and motor skills are delayed with a mean age of walking of four to six years (range 27 months – 7 years); some affected individuals may walk only with assistance and others do not acquire independent walking skills [Whalen et al 2012]. Those who walk independently often have a wide-based, unsteady gait. Most require orthotics to stabilize the ankle.
Speech is significantly delayed in all individuals with PTHS and excessive drooling is common. Most are nonverbal. Some have developed a few words, but subsequently regressed later in life and become entirely nonverbal. Rare individuals can string words into sentences. Receptive language is generally stronger than expressive language, and many individuals with PTHS are able to understand and follow simple commands. The use of augmentative communication devices has been useful for many individuals, though other individuals lack the fine motor skills to make use of these devices.
Self-care skills are also delayed and few are reported to develop consistent dressing or toileting skills.
Behavioral. PTHS is most commonly associated with autism spectrum disorder (ASD) symptoms, characterized by impairments in communication, behavior, and social interactions. The relationship between autism and PTHS remains ambiguous; however, in a study conducted by Van Balkom et al, the Autism Diagnostic Interview Revised (ADI-R) assessed ten participants with confirmed PTHS diagnoses who scored at or above social, behavioral, and communication criteria for ASD (2 participants did not score above the behavioral domain cutoff value). Six out of ten participants were observed to have fixations that prompted repetitive exposure to a specific object, listening to the same song, or viewing the same video repeatedly [Van Balkom et al 2012].
Many individuals with PTHS are described as having a happy disposition [Zweier et al 2008, Marangi et al 2011], though it has been proposed that an anxious disposition with a smiling appearance is a more apt description [Whalen et al 2012]. Others are described as being difficult to handle as they develop outbursts of aggression or bouts of shouting associated with frustration or unanticipated changes in routine [Andrieux et al 2008, Giurgea et al 2008, de Pontual et al 2009]. The onset of puberty can be associated with increased aggression and behavioral issues [Authors, personal observation].
Individuals with PTHS may be shy or anxious in new situations and self-aggression can occur. Disruptions of routine have also been associated with episodes of anxiety. Hand biting and head banging have been commonly reported by families of individuals with PTHS.
Unprovoked laughter may occur. Sleep disturbance in childhood is reported in fewer than half of affected individuals [Whalen et al 2012] and includes difficulty falling asleep, problems sleeping through the night, and night terrors [de Winter et al 2016].
Stereotypic head movements (e.g., head rotation) and stereotypic hand movements (e.g., flapping, clapping, washing movements, hand to mouth, finger crossing) are commonly observed [Takano et al 2010, Marangi et al 2011, Whalen et al 2012]. Three individuals have been reported to have lost the use of hand skills [Zweier et al 2008, Armani et al 2012, Whalen et al 2012].
Individuals with PTHS invariably have a profound affinity for music. Families report that during episodes of anxiety or frustration, music will very often soothe or placate them. Many appear to love playing with water, and exhibit a fascination with running water [Authors, personal observation].
Craniofacial. In addition to the craniofacial features discussed in Diagnosis, cherry red lips have been noted infrequently [Ghosh et al 2012].
Growth. Growth is typically in the normal range for size at birth; slower postnatal growth is noted in about 25%.
Head growth was found to slow postnatally in 26% [de Winter et al 2016], with microcephaly reported in up to 60% [Zweier et al 2008, Goodspeed et al 2018].
The initiation and progression of secondary sexual characteristics appear to be unimpaired in individuals with PTHS [Authors, personal observation].
Respiratory. Episodic hyperventilation often followed by breath-holding/apnea while awake are reported in 40%-60% of individuals [Whalen et al 2012, de Winter et al 2016, Goodspeed et al 2018]. Hyperventilation is often associated with anxiety or excitement [Giurgea et al 2008, Steinbusch et al 2013] and is not present during sleep [Maini et al 2012]. Breath-holding may be associated with cyanosis, and in rare cases syncope, and can occur independently of hyperventilation [Whalen et al 2012]. Finger clubbing and chronic hypoxia has been noted in 7%-19% [Whalen et al 2012, Goodspeed et al 2018].
Breathing abnormalities tend to develop between ages three and seven years; few individuals are affected at birth. In one study 30% had symptoms by age 15 years [de Winter et al 2016]. In some children, hyperventilation and/or breath-holding episodes are observed for only a few months or years. However, in most affected individuals breathing abnormalities persist to some degree, though too few adults have been described to make conclusive statements about frequency or severity. Breath-holding/apnea episodes are not typically related to seizure activity [Maini et al 2012]. Treatment can mitigate the severity and frequency of symptoms.
Neurologic. Seizures, reported in 40%-50% of individuals with PTHS, vary in type and severity. Onset of seizures is reported from early infancy to as late as age 18 years [de Pontual et al 2009].
In an internet questionnaire study of 101 families, 37.6% had epilepsy [de Winter et al 2016]. Of those with a history of epilepsy, 23.7% became seizure-free at a mean age of 6.4 years. The majority (51.4%) had involuntary motor movements or tonic-clonic seizures often with absence seizures, 16.2% had only absence seizures, and the rest could not accurately describe their family member's seizures. Since this was a questionnaire study, it is unclear whether "absence" events were true absence seizures or focal-onset seizures with no motor component.
The breathing abnormality in PTHS does not appear to be a manifestation of seizure activity [Maini et al 2012], though in this study, 7/38 individuals with seizures had apnea or hyperventilation shortly before their seizures.
In most individuals with seizures, these are well controlled by antiepileptic drugs – valproic acid, levetiracetam, lamotrigine, and carbamazepine being the most common [de Winter et al 2016].
EEG and MRI findings reported in PTHS are limited.
- One study reported EEGs in four individuals with PTHS; no specific patterns other than occipital and central delta waves were found in the younger two individuals. Pseudoperiodic complexes were present during wakefulness over the central and occipital regions, at times admixed with slow spike and wave in the two older individuals [Amiel et al 2007].
- In those individuals who have had a brain MRI, a variety of findings have been reported, though many studies are reported as normal. The most common findings include white matter abnormalities such as hypoplasia/agenesis of the corpus callosum and white matter hyperintensity in the temporal poles, as well as ventricular dilatation, posterior fossa abnormalities, small hippocampi, and moderate hypoplasia of the frontal lobes [Amiel et al 2007, Marangi et al 2011, Whalen et al 2012].
Signs of general autonomic dysfunction have been observed in some individuals or reported by families [Authors, personal observation]. These include:
- High pain tolerance (very common);
- Lack of tears;
- Impaired ability to sweat and dysregulation of body temperature homeostasis.
Eyes. Myopia, strabismus, and/or astigmatism are present in 50%-60%. Myopia can be severe (>6 diopters) and evident before age two years [Giurgea et al 2008, Stavropoulos et al 2010].
Gastrointestinal. Early feeding issues may occur, although most resolve with age.
Constipation is common (75%) and may be severe. Hirschsprung disease is rare, with one individual reported [Peippo et al 2006].
Gastroesophageal reflux is reported in fewer than half of individuals with PTHS [de Winter et al 2016]. In some individuals, reflux is associated with symptoms of impaired swallowing, and videofluoroscopic swallowing studies may be useful.
Musculoskeletal. Minor hand and foot anomalies such as slender or small hands and feet, broad fingertips, clinodactyly, tapered fingers, transverse palmar crease, flat feet with hindfoot valgus deformity, overriding toes, and short metatarsals have been reported. Absent flexion creases of the thumbs may occur with thumb ankylosis. In one individual an absent thumb tendon was found during surgery [Authors, personal observation].
Hands and feet are reported to be cold and cyanosed in some individuals.
Hyperpronation of the ankles is a nearly universal feature.
Clubfeet have been reported in 20% [Whalen et al 2012].
Scoliosis has been noted in about 25% although no information regarding severity is available.
Skin. Many have had prominent pads on the fingertips and/or toes (persistent fetal pads) [Lehalle et al 2011].
Supernumerary nipples have been observed in at least ten individuals [Rosenfeld et al 2009, Takano et al 2010, Marangi et al 2011].
Adult-onset features. Relatively few older teens or adults have been reported with PTHS; thus it is not yet known what, if any, other adult-onset findings may be of concern in PTHS. Several adults have had significant hand tremors. Another had chronic urinary retention requiring intermittent catheterization [Authors, personal observations].
Other. Hodgkin lymphoma has been reported in an individual age 29 years with PTHS [Zweier et al 2007]. While TCF4 has a role in lymphocyte development [Zhuang et al 1996], the lack of other individuals with PTHS reported to have lymphoma suggests that this finding is coincidental.
Genotype-Phenotype Correlations
According to most studies reported, the phenotype associated with PTHS appears to be independent of TCF4 variant type.
Size of deletion. In several studies comparing chromosomal deletions of varying lengths of chromosome 18q21.2 involving TCF4, individuals appeared to have similar phenotypes to each other and single-nucleotide variants in TCF4 [Andrieux et al 2008, Hasi et al 2011, Stavropoulos et al 2010, Giurgea et al 2008]. However, one case suggested that haploinsufficiency of the neighboring genes MBD1 and MBD2 may confer a more severe Rett syndrome-like phenotype [Kato et al 2010].
Mosaicism for chromosome 18q21.1 deletions have been described. Two individuals with mosaicism for chromosome 18q21.2 deletions (11.9 Mb and 7.5 Mb in size) demonstrated the typical PTHS phenotype [Giurgea et al 2008, Stavropoulos et al 2010]. Another individual with mosaic 18q21.2 deletion (185 kb) involving a portion of TCF4 was reported to have dysmorphic features, attention-deficit disorder, anxiety, self-mutilation, and stiff joints [Pham et al 2014]. Two other individuals mosaic for 18q21 deletions including TCF4 have been described with dysmorphic facial features (though not classic for PTHS), severe developmental delay, and microcephaly. One of these individuals had 5%-10% mosaicism and the other about 40% [Rossi et al 2012].
Penetrance
Penetrance of a pathogenic TCF4 allele is complete, though some phenotypic variability is seen among sibs born to a presumably mosaic parent.
Prevalence
Overall prevalence of PTHS is unknown. However, based on a lower relative detection rate as compared to Williams syndrome and Smith-Magenis syndrome deletions, one laboratory estimated that the frequency of chromosome 18q21 deletions associated with PTHS is between 1:34,000 and 1:41,000 [Rosenfeld et al 2009]. If deletions are found in approximately one third of individuals with PTHS, the frequency of the condition could be as high as 1:11,000.
PTHS occurs in both males and females and is not limited to any specific ethnic background.
Genetically Related (Allelic) Disorders
Trinucleotide repeat expansion in TCF4 has been associated with Fuchs endothelial corneal dystrophy [Wieben et al 2017].
Studies suggest that TCF4 pathogenic variants are involved in an unrecognized subset of those with nonsyndromic intellectual disabilities of varying severity [Kalscheuer et al 2008, Kharbanda et al 2016].
Differential Diagnosis
See Table 2 for disorders with features that overlap those of Pitt-Hopkins syndrome (PTHS).
Table 2.
Disorders to Consider in the Differential Diagnosis of Pitt-Hopkins Syndrome
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with Pitt-Hopkins syndrome (PTHS), the following evaluations are recommended if they have not already been completed:
- Developmental assessment to establish a cognitive baseline and determine the types of services and educational strategies needed. In school-aged children this information is important for the child's individualized education plan (IEP).
- Assessment for the use of nonverbal communication devices and strategies (given that most individuals do not develop useful speech)
- Child behavior specialist to aid with behavioral concerns. Consultation with a child psychiatrist may be warranted if there are significant behavioral issues.
- Pulmonary consultation if history of respiratory pattern abnormality is identified on directed questioning. Polysomnography may be indicated if there is a history of episodic apnea, especially when associated with cyanosis (typically alternating with episodes of hyperventilation).
- Child neurology evaluation to establish a neurologic baseline and evaluate for other neurologic issues such as sleep dysfunction or seizures. This should be expedited if there are familial concerns about seizure activity. Consider a baseline polysomnogram to assess for sleep disturbances.
- Ophthalmology evaluation to evaluate for myopia, astigmatism, and/or strabismus
- Gastroenterology evaluation to establish treatment regimen for chronic constipation and other possible GI issues such as gastroesophageal reflux
- Musculoskeletal evaluation by an orthopedist or physiatrist to evaluate ambulatory skills and need for special mobility equipment and/or orthotics to aid in foot position
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an IEP is developed.
Ages 5-21 years
- In the US, an IEP based on the individual's level of function should be developed by the local public school district. Affected children are permitted to remain in the public school district until age 21.
- Discussion about transition plans including financial, vocation/employment, and medical arrangements should begin at age 12 years. Developmental pediatricians can provide assistance with transition to adulthood.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, Botox®, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction. Assuming that the individual is safe to eat by mouth, feeding therapy – typically from an occupational or speech therapist – is recommended for affected individuals who have difficulty feeding due to poor oral motor control.
Communication issues. Strong consideration should be given to early training with alternative means of communication (e.g., Augmentative and Alternative Communication [AAC]) for those with PTHS, given the presence of severe expressive language difficulties.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and is typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Other Issues
Pulmonary. Some reports indicate that antiepileptic medications control seizures while leaving the unusual respiratory patterns unchanged [Peippo et al 2006]; others have noted some decrease in the frequency of the respiratory episodes with the use of anticonvulsants [Takano et al 2010].
- Improvement of an abnormal respiratory pattern after treatment with sodium valproate has been reported in a person with PTHS with frequent apneic episodes associated with hypoxemia [Maini et al 2012].
- Case reports of individuals with PTHS demonstrated that daily treatment with acetazolamide resulted in decreased frequency and duration of hyperventilatory and apneic episodes and improved oxygen saturation [Verhulst et al 2012, Gaffney & McNally 2015].Those on acetazolamide should have routine monitoring of electrolytes and acid-base status.
Neurologic. Treatment of epilepsy should be tailored to the type of seizure and individualized to the needs of the affected person. If present, other neurologic issues such as sleep dysfunction should be treated.
Ophthalmologic. Eyeglasses or surgery are indicated as needed for amblyopia. Most, if not all, individuals with PTHS may exhibit a form of refractive error. These also may appear at any age, and as such there may be a benefit in regular ophthalmologic exams in people with PTHS.
Gastrointestinal. In most individuals, regular use of high-fiber diet and/or laxative regimen to address constipation and antacids for reflux are appropriate.
Musculoskeletal. Nearly all affected individuals require orthotics for abnormal foot position to aid in ambulation. Orthopedic treatment of scoliosis and other musculoskeletal dysfunction as needed is appropriate.
Other. Standard care is indicated for other medical issues.
Surveillance
Appropriate surveillance includes:
- Ongoing developmental assessments to tailor educational services to an individual's strengths;
- Regular follow up with an ophthalmologist to monitor for high myopia and strabismus;
- Periodic reevaluation with clinical geneticist and/or genetic counselor to review the most current information and recommendations for individuals with PTHS.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Pitt-Hopkins syndrome (PTHS) is caused by haploinsufficiency of TCF4 resulting from either a pathogenic variant in TCF4 or a deletion of the chromosome region in which TCF4 is located (18q21.2). Most individuals reported to date have been simplex cases (i.e., a single occurrence in a family) resulting from a de novo pathogenic variant or deletion.
Risk to Family Members – TCF4 Pathogenic Variant
Parents of a proband
- Most individuals with PTHS reported to date have the disorder as the result of a de novo genetic alteration and both parents are unaffected.
- Molecular genetic testing is recommended for the parents of a proband.
- If the TCF4 pathogenic variant identified in the proband cannot be identified in the leukocyte DNA of either parent, the most likely explanation is that the pathogenic variant is de novo in the proband. Another possible explanation is germline and/or somatic and germline mosaicism in a parent.
- Somatic and (presumed) germline mosaicism has been identified in at least two parents:
- The unaffected mother of two sibs diagnosed with PTHS [Steinbusch et al 2013]
- The mother (of an affected child) who had been treated for chronic depression and epilepsy since age 20 years [de Pontual et al 2009]
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If the parents have been tested for the TCF4 pathogenic variant identified in the proband and:
- A parent of the proband has the TCF4 pathogenic variant (even in a mosaic state), the risk to the sibs of inheriting the variant could be as high as 50%. Sibs who inherit a TCF4 pathogenic variant will be affected; phenotypic variability among affected sibs (born to a presumably mosaic parent) has been reported [Steinbusch et al 2013; Author, personal observation].
- If the TCF4 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism. Parental germline mosaicism for PTHS has been inferred [de Pontual et al 2009, Steinbusch et al 2013].
- If the parents have not been tested for the TCF4 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, the sibs of a proband with clinically unaffected parents are still at increased risk for PTHS because of the possibility of parental germline mosaicism.
Offspring of a proband. There have been no reports of individuals with PTHS reproducing.
Other family members. Because PTHS typically occurs as a de novo genetic alteration, the risk to other family members is presumed to be low.
Risk to Family Members – Chromosome 18q21.2 Deletion
Parents of a proband
- The chromosome 18q21.2 deletion (whether resulting from an "apparently balanced" chromosome rearrangement or, more commonly, from a simple deletion) has been de novo in almost all affected individuals (reported in the medical literature) whose parents had cytogenetic studies.
- Parents of a proband are typically not affected.
- Rare instances of somatic mosaicism have been detected in a parent of an affected individual. One of these parents had chronic depression and epilepsy from age 20 years; the other was healthy [Steinbusch et al 2013, de Pontual et al 2009].
- Evaluation of the parents by genomic testing that will detect the chromosome 18q21.2 deletion present in the proband may be offered (note: low-level somatic and/or germline mosaicism in a parent can never be entirely ruled out by testing).
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a chromosome 18q21.2 deletion identified in the proband is not identified in one of the parents, the risk to sibs is slightly greater than that of the general population (though still <1%) because of the possibility of parental germline mosaicism for the deletion.
- If a proband's parent has a chromosome 18q21.2 deletion (even in a mosaic state), the risk for sibs to be similarly affected could be as high as 50%.
Offspring of a proband. There have been no reports of individuals with PTHS reproducing.
Other family members. Because PTHS typically occurs as a de novo genetic alteration, the risk to other family members is presumed to be low.
Related Genetic Counseling Issues
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the variant identified in the proband or clinical evidence of the disorder, the variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of affected individuals.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of affected individuals.
Prenatal Testing and Preimplantation Genetic Testing
Once the PTHS-related genetic alteration has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- National Organization for Rare Disorders (NORD)
- Pitt Hopkins Research FoundationThe mission of the Pitt Hopkins Research Foundation (PHRF) is to support research dedicated to finding a treatment, and an eventual cure of Pitt Hopkins syndrome and other similar disorders. The PHRF is also dedicated to supporting the Pitt Hopkins community with resource recommendations, parental support and the latest medical information.
- Chromosome 18 Registry and Research SocietyPhone: 210-657-4968Email: Office@Chromosome18.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Pitt-Hopkins Syndrome: Genes and Databases
Table B.
OMIM Entries for Pitt-Hopkins Syndrome (View All in OMIM)
Gene structure. TCF4 has 18 coding exons (exons 2-19) spanning 360 kb with exons 1 and 20 being noncoding. However, a bioinformatic analysis of mRNA and expressed sequence tag sequences demonstrates that TCF4 uses numerous alternative 5' exons, potentially yielding many isoforms with different N-termini and different subcellular distribution [Sepp et al 2011]. TCF4 mRNAs are ubiquitously expressed but the levels differ considerably between different tissues, with the highest levels present in fetal brain [Sepp et al 2011]. See Table A, Gene for a detailed summary of gene and protein information.
Pathogenic variants. More than 130 PTHS-associated pathogenic variants have been described in the literature to date; with the exception of five recurrent variants, all are private. Whalen et al [2012] summarized the frequency of various types of TCF4 pathogenic variants:
- 30% are deletions including one or more exons and varying in size from a single exon to several megabases. Deletions result in either a frameshift or removal of the basic helix-loop-helix (bHLH) domain.
- 30% are small intra-exon insertions or deletions.
- 40% are single-nucleotide variants:
- Nonsense variants account for 20% of all single-nucleotide variants reported and are spread throughout the gene between exons 7 and 18. They are presumed to result in nonsense-mediated RNA decay.
- Splice variants account for 14% of single-nucleotide variants and affect mostly donor or acceptor consensus sites. Splice variants in TCF4 are thought to result in frameshifts, and no splice variants affecting exon 15 (the only in-frame exon) have been reported to date.
- Pathogenic missense variants represent 19% of single-nucleotide variants in TCF4 and mostly affect conserved residues in the bHLH domain, several of which have been seen in multiple families. Rare pathogenic variants have been reported in the AD2 and repression domains. There is little evidence to support the pathogenicity of missense variants outside these domains.
- A hot spot in the nucleotides encoding the basic domain of TCF4 accounts for 25% of pathogenic variants [Whalen et al 2012].
- The penetrance of a pathogenic TCF4 allele is apparently complete.
A genotype-phenotype correlation has been proposed based on the position of pathogenic variants in TCF4, with deletions involving the 5' end of TCF4 producing a milder phenotype. Less severe pathogenic variants that may result in milder or atypical presentations include the following:
- A family with deletion of exons 1-4 of TCF4 and mild-to-moderate intellectual disability without the characteristic phenotype of Pitt-Hopkins syndrome (PTHS) was described by Kharbanda et al [2016].
- An individual age 55 years with deletion of exons 4 to 6 of TCF4 and some facial features of PTHS but only mild intellectual delay was described by Bedeschi et al [2017].
- An individual with mild-to-moderate intellectual disability and minor facial abnormalities but without classic PTHS was reported to have a balanced translocation t(18:20)(q21.1;q11.2) disrupting TCF4 upstream to exon 4. Fusion transcripts between TCF4 and CHD6 produced in the affected individual's cell line may result in a partially functional TCF4 protein [Kalscheuer et al 2008].
- Segregation of mild intellectual disability with a complex unbalanced translocation disrupting TCF4 in a three-generation family and the characterization of resultant transcripts suggested that the overexpression of short isoforms can partially rescue the Pitt-Hopkins phenotype [Maduro et al 2016].
In contrast, nonsense or frameshift pathogenic variants in exons 7 and 8 appeared to result in more severe intellectual disability with less distinctive facial features, while pathogenic variants in exons 9 to 19 resulted in classic PTHS [Bedeschi et al 2017]. These observations are limited by the scarcity of individuals described in the literature with pathogenic variants in exons 1 to 9 and the reliance on incomplete phenotypic descriptions.
Genetic and epigenetic modifiers as well as environmental factors may also influence the phenotype. Two brothers with the same TCF4 pathogenic variant demonstrated different severity of phenotype: one brother showed the typical characteristics of PTHS; the other brother had milder physical features and less severe intellectual disability. Their unaffected mother was somatic mosaic for the TCF4 pathogenic variant [Steinbusch et al 2013].
Normal gene product. TCF4 encodes a member of the class I basic helix-loop-helix (bHLH) family of transcription factors, which regulate multiple processes including cellular differentiation and proliferation and lineage commitment [Atchley & Fitch 1997, Kageyama & Nakanishi 1997, Ross et al 2003, de Pontual et al 2009]. During early development, TCF4 is highly expressed in the central nervous system, genital bud, peribronchial and kidney mesenchyme, and sclerotome [de Pontual et al 2009]. Homozygous Tcf4 knockout mice exhibit early lethality, suggesting an important role in development [Zhuang et al 1996].
Class I bHLH proteins are also known as E-proteins because they bind DNA as homo- or heterodimers, at Ephrussi box (E-box) sequences (CANNTG) [Ephrussi et al 1985, Massari & Murre 2000]. E-proteins have two conserved transactivation domains, AD1 and AD2 [Aronheim et al 1993, Quong et al 1993], a repression domain, RD [Markus et al 2002], as well as a common bHLH structural motif that mediates homo- and heterodimerization between bHLH proteins via their HLH domain, while the adjacent basic region mediates the DNA binding to the E-box [Ross et al 2003]. The interaction of TCF4 with other bHLH proteins indicates a potential role in development of specific parts of the central and peripheral nervous system.
Several TCF4 isoforms have been reported, the longest of which encodes a protein of 671 amino acids [Sepp et al 2011]. See Table A.
Abnormal gene product. PTHS is caused by haploinsufficiency of TCF4 protein [Amiel et al 2007]. The clinical features observed in individuals with PTHS are consistent with haploinsufficiency of TCF4 having a negative effect on development, particularly affecting specific parts of the central and peripheral nervous system [Persson et al 2000].
Tcf4 knockout mice have disrupted development of the pontine nucleus [Flora et al 2007]. TCF4-haploinsufficient mice exhibit defects in behavior and memory that can be treated with HDAC inhibition [Kennedy et al 2016]. Converging results from analysis of a mouse model of Tcf4 knockdown in the developing mouse frontal cortex and of a mouse model with a truncated Tcf4 allele show upregulation of the Scn10a ion channel as a potential therapeutic target for Pitt-Hopkins syndrome [Rannals et al 2016].
References
Literature Cited
- Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, Plouin P, Carter NP, Lyonnet S, Munnich A, Colleaux L. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80:988–93. [PMC free article: PMC1852736] [PubMed: 17436254]
- Andrieux J, Lepretre F, Cuisset JM, Goldenberg A, Delobel B, Manouvrier-Hanu S, Holder-Espinasse M. Deletion 18q21.2q21.32 involving TCF4 in a boy diagnosed by CGH-array. Eur J Med Genet. 2008;51:172–7. [PubMed: 18222743]
- Armani R, Archer H, Clarke A, Vasudevan P, Zweier C, Ho G, Williamson S, Cloosterman D, Yang N, Christodoulou J. Transcription factor 4 and myocyte enhancer factor 2C mutations are not common causes of Rett syndrome. Am J Med Genet A. 2012;158A:713–9. [PubMed: 22383159]
- Aronheim A, Shiran R, Rosen A, Walker MD. The E2A gene product contains two separable and functionally distinct transcription activation domains. Proc Natl Acad Sci U S A. 1993;90:8063–7. [PMC free article: PMC47288] [PubMed: 8367464]
- Atchley WR, Fitch WM. A natural classification of the basic helix-loop-helix class of transcription factors. Proc Natl Acad Sci U S A. 1997;94:5172–6. [PMC free article: PMC24651] [PubMed: 9144210]
- Bedeschi MF, Marangi G, Calvello MR, Ricciardi S, Leone FPC, Baccarin M, Guerneri S, Orteschi D, Murdolo M, Lattante S, Frangella S, Keena B, Harr MH, Zackai E, Zollino M. Impairment of different protein domains causes variable clinical presentation within Pitt-Hopkins syndrome and suggests intragenic molecular syndromology of TCF4. Eur J Med Genet. 2017;2017;60:565–71. [PubMed: 28807867]
- Brockschmidt A, Todt U, Ryu S, Hoischen A, Landwehr C, Birnbaum S, Frenck W, Radlwimmer B, Lichter P, Engels H, Driever W, Kubisch C, Weber RG. Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum Mol Genet. 2007;16:1488–94. [PubMed: 17478476]
- de Pontual L, Mathieu Y, Golzio C, Rio M, Malan V, Boddaert N., Soufflet C, Picard C, Durandy A, Dobbie A, Heron D, Isidor B, Motte J, Newburry-Ecob R, Pasquier L, Tardieu M, Viot G, Jaubert F, Munnich A, Colleaux L, Vekemans M, Etchevers H, Lyonnet S, Amiel J. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat. 2009;30:669–76. [PubMed: 19235238]
- de Winter CF, Baas M, Bijlsma EK, Heukelingen JV, Routledge S, Hennekam CM. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis. 2016;11:37. [PMC free article: PMC4830011] [PubMed: 27072915]
- Ephrussi A, Church GM, Tonegawa S, Gilbert W. B lineage--specific interactions of an immunoglobulin enhancer with cellular factors in vivo. Science. 1985;227:134–40. [PubMed: 3917574]
- Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-protein Tcf4 interacts with Math 1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci U S A. 2007;104:15382–7. [PMC free article: PMC1978485] [PubMed: 17878293]
- Gaffney C, McNally P. Successful use of acetazolamide for central apnea in a child with Pitt-Hopkins syndrome. Am J Med Genet A. 2015;167:1423. [PubMed: 25900839]
- Ghosh PS, Freidman NR, Ghosh D. Pitt-Hopkins syndrome in a boy with Charcot Marie Tooth disease type 1A: a rare co-occurrence of 2 genetic disorders. J Child Neurol. 2012;27:1602–6. [PubMed: 22378661]
- Giurgea I, Missirian C, Cacciagli P, Whalen S, Fredriksen T, Gaillon T, Rankin J, Mathieu-Dramard M, Morin G, Martin-Coignard D, Dubourg C, Chabrol B, Arfi J, Giuliano F, Claude Lambert J, Philip N, Sarda P, Villard L, Goossens M, Moncla A. TCF4 deletions in Pitt-Hopkins Syndrome. Hum Mutat. 2008;29:E242–51. [PubMed: 18781613]
- Goodspeed K, Newsom C, Morris MA, Powell C, Evans P, Golla S. Pitt-Hopkins Syndrome: A Review of Current Literature, Clinical Approach, and 23-Patient Case Series. J Child Neurol. 2018;33:233–44. [PMC free article: PMC5922265] [PubMed: 29318938]
- Hasi M, Soileau B, Sebold C, Hill A, Hale DE, O'Donnell L, Cody JD. The role of the TCF4 gene in the phenotype of individuals with 18q segmental deletions. Hum Genet. 2011;130:777–87. [PMC free article: PMC3215814] [PubMed: 21671075]
- Kageyama R, Nakanishi S. Helix-loop-helix factors in growth and differentiation of the vertebrate nervous system. Curr Opin Genet Dev. 1997;7:659–65. [PubMed: 9388783]
- Kalscheuer VM, Feenstra I, Van Ravenswaaij-Arts CM, Smeets DF, Menzel C, Ullmann R, Musante L, Ropers HH. Disruption of the TCF4 gene in a girl with mental retardation but without the classical Pitt-Hopkins syndrome. Am J Med Genet A. 2008;146A:2053–9. [PubMed: 18627065]
- Kato Z, Morimoto W, Kimura T, Matsushima A, Kondo N. Interstitial deletion of 18q: comparative genomic hybridization array analysis of 46, XX,del(18)(q21.2.q21.33). Birth Defects Res A Clin Mol Teratol. 2010;88:132–5. [PubMed: 19813260]
- Kennedy AJ, Rahn EJ, Paulukaitis BS, Savell KE, Kordasiewicz HB, Wang J, Lewis JW, Posey J, Strange SK, Guzman-Karlsson MC, Phillips SE, Decker K, Motley ST, Swayze EE, Ecker DJ, Michael TP, Day JJ, Sweatt JD. Tcf4 Regulates Synaptic Plasticity, DNA Methylation, and Memory Function. Cell Rep. 2016;16:2666–85. [PMC free article: PMC5710002] [PubMed: 27568567]
- Kharbanda M, Kannike K, Lampe A, Berg J, Timmusk T, Sepp M. Partial deletion of TCF4 in three generation family with non-syndromic intellectual disability, without features of Pitt-Hopkins syndrome. Eur J Med Genet. 2016;59:310–4. [PubMed: 27132474]
- Lehalle D, Williams C, Siu VM, Clayton-Smith J. Fetal pads as a clue to the diagnosis of Pitt-Hopkins syndrome. Am J Med Genet A. 2011;155A:1685–9. [PubMed: 21671383]
- Maduro V, Pusey BN, Cherukuri PF, Atkins P, du Souich C, Rupps R, Limbos M, Adams DR, Bhatt SS, Eydoux P, Links AE, Lehman A, Malicdan MC, Mason CE, Morimoto M, Mullikin JC, Sear A, Van Karnebeek C, Stankiewicz P, Gahl WA, Toro C, Boerkoel CF. Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability. Orphanet J Rare Dis. 2016;11:62. [PMC free article: PMC4868023] [PubMed: 27179618]
- Maini I, Cantalupo G, Turco EC, De Paolis F, Magnani C, Parrino L, Terzano MG, Pisani F. Clinical and Polygraphic Improvement of Breathing Abnormalities After Valproate in a Case of Pitt-Hopkins Syndrome. J Child Neurol. 2012;27:1585–8. [PubMed: 22378662]
- Marangi G, Ricciardi S, Orteschi D, Lattante S, Murdolo M, Dallapiccola B, Biscione C, Lecce R, Chiurazzi P, Romano C, Greco D, Pettinato R, Sorge G, Pantaleoni C, Alfei E, Toldo I, Magnani C, Bonanni P, Martinez F, Serra G, Battaglia D, Lettori D, Vasco G, Baroncini A, Daolio C, Zollino M. The Pitt-Hopkins syndrome: report of 16 new patients and clinical diagnostic criteria. Am J Med Genet A. 2011;155A:1536–45. [PubMed: 21671391]
- Markus M, Du Z, Benezra R. Enhancer-specific modulation of E protein activity. J Biol Chem. 2002;277:6469–77. [PubMed: 11724804]
- Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429–40. [PMC free article: PMC85097] [PubMed: 10611221]
- Peippo MM, Simola KO, Valanne LK, Larsen AT, Kähkönen M, Auranen MP, Ignatius J. Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clin Dysmorphol. 2006;15:47–54. [PubMed: 16531728]
- Persson P, Jogi A, Grynfeld A, Pahlman S, Axelson H. HASH-1 and E2-2 are expressed in human neuroblastoma cells and form a functional complex. Biochem Biophys Res Commun. 2000;274:22–31. [PubMed: 10903890]
- Pham J, Shaw C, Pursley A, Hixson P, Sampath S, Roney E, Gambin T, Kang S-HL, Bi W, Lalani S, Bacino C, Lupski JR, Stankiewicz P, Patel A, Cheung S-W. Somatic mosaicism detected by exon-targeted, high-resolution aCGH in 10,362 consecutive cases. Eur J Hum Genet. 2014;22:969–78. [PMC free article: PMC4350600] [PubMed: 24398791]
- Quong MW, Massari ME, Zwart R, Murre C. A new transcriptional-activation motif restricted to a class of helix-loop-helix proteins is functionally conserved in both yeast and mammalian cells. Mol Cell Biol. 1993;13:792–800. [PMC free article: PMC358962] [PubMed: 8423802]
- Rannals MD, Page SC, Campbell MN, Gallo RA, Mayfield B, Maher BJ. Neurodevelopmental models of transcription factor 4 deficiency converge on a common ion channel as a potential therapeutic target for Pitt Hopkins syndrome. Rare Dis. 2016;4:e1220468. [PMC free article: PMC5154382] [PubMed: 28032012]
- Rosenfeld JA, Leppig K, Ballif BC, Thiese H, Erdie-Lalena C, Bawle E, Sastry S, Spence JE, Bandholz A, Surti U, Zonana J, Keller K, Meschino W, Bejjani BA, Torchia BS, Shaffer LG. Genotype-phenotype analysis of TCF4 mutations causing Pitt-Hopkins syndrome shows increased seizure activity with missense mutations. Genet Med. 2009;11:797–805. [PubMed: 19938247]
- Ross SE, Greenberg ME, Stiles CD. Basic helix-loop-helix factors in cortical development. Neuron. 2003;39:13–25. [PubMed: 12848929]
- Rossi M, Labalme A, Cordier MP, Till M, Blanchard G, Dubois R, Guibaud L, Heissat S, Javouhey E, Lachaux A, Mure PY, Ville D, Edery P, Sanlaville D. Mosaic 18q21.2 deletions including the TCF4 gene: a clinical report. Am J Med Genet A. 2012;158A:3174–81. [PubMed: 23165966]
- Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5' exon usage and splicing. PLoS One. 2011;6:e22138. [PMC free article: PMC3137626] [PubMed: 21789225]
- Stavropoulos DJ, MacGregor DL, Yoon G. Mosaic microdeletion 18q21 as a cause of mental retardation. Eur J Med Genet. 2010;53:396–9. [PubMed: 20813211]
- Steinbusch CV, van Roozendaal K, Tserpelis D, Smeets E, Kranenburg-de Koning T, de Waal K, Zweier C, Rauch A, Hennekam R, Blok M, Schrander-Stumpel C. Somatic mosaicism in a mother of two children with Pitt-Hopkins syndrome. Clin Genet. 2013;83:73–7. [PubMed: 22335494]
- Takano K, Lyons M, Moyes C, Jones J, Schwartz CE. Two percent of patients suspected of having Angelman syndrome have TCF4 mutations. Clin Genet. 2010;78:282–8. [PubMed: 20184619]
- Takano K, Tan WH, Irons MB, Jones JR, Schwartz CE. Pitt-Hopkins syndrome should be in the differential diagnosis for males presenting with an ATR-X phenotype. Clin Genet. 2011;80:600–1. [PubMed: 22040220]
- Van Balkom ID, Vuijk PJ, Franssens M, Hoek HW, Hennekam RC. Development, cognition, and behavior in Pitt-Hopkins syndrome. Dev Med Child Neurol. 2012;54:925–31. [PubMed: 22712893]
- Verhulst SL, DeDooy J, Ramet J, Bockaert N, VanCoster R, Ceulemans B, DeBacker W. Acetazolamide for severe apnea in Pitt-Hopkins syndrome. Am J Med Genet. 2012;158A:932–4. [PubMed: 22407847]
- Whalen S, Héron D, Gaillon T, Moldovan O, Rossi M, Devillard F, Giuliano F, Soares G, Mathieu-Dramard M, Afenjar A, Charles P, Mignot C, Burglen L, Van Maldergem L, Piard J, Aftimos S, Mancini G, Dias P, Philip N, Goldenberg A, Le Merrer M, Rio M, Josifova D, Van Hagan JM, Lacombe D, Edery P, Dupuis-Girod S, Putoux A, Sanlaville D, Fischer R, Drévillon L, Briand-Suleau A, Metay C, Goossens M, Amiel J, Jacquette A, Giurgea I. Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: Clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat. 2012;33:64–72. [PubMed: 22045651]
- Wieben ED, Aleff RA, Tang X, Butz ML, Kalari KR, Highsmith EW, Jen J, Vasmatzis G, Patel SV, Maguire LJ, Baratz KH, Fautsch MP. Trinucleotide repeat expansion in the transcription factor 4 (TCF4) gene leads to widespread mRNA splicing changes in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2017;58:343–52. [PMC free article: PMC5270622] [PubMed: 28118661]
- Zhuang Y, Cheng P, Weintraub H. B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2-2, and HEB. Mol Cell Biol. 1996;16:2898–905. [PMC free article: PMC231283] [PubMed: 8649400]
- Zweier C, Peippo MM, Hoyer J, Sousa S., Bottani A, Clayton-Smith J, Reardon W, Saraiva J, Cabral A, Gohring I, Devriendt K, de Ravel T, Bijlsma EK, Hennekam RC, Orrico A, Cohen M, Dreweke A, Reis A, Nurnbert P, Rauch A. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am J Hum Genet. 2007;80:994–1001. [PMC free article: PMC1852727] [PubMed: 17436255]
- Zweier C, Sticht H, Bijlsma EK, Clayton-Smith J, Boonen SE, Fryer A, Greally MT, Hoffmann L, den Hollander NS, Jongmans M, Kant SG, King MD, Lynch SA, McKee S, Midro AT, Park SM, Ricotti V, Tarantino E, Wessels M, Peippo M, Rauch A. Further delineation of Pitt-Hopkins syndrome: phenotypic and genotypic description of 16 novel patients. J Med Genet. 2008;45:738–44. [PubMed: 18728071]
Chapter Notes
Author History
Holly H Ardinger, MD; University of Missouri-Kansas City (2012-2018)
Ibrahim Elsharkawi, MD (2018-present)
Kimberly Parkin (2018-present)
Carol J Saunders, PhD; University of Missouri-Kansas City (2012-2018)
Marci Steeves, MS, CGC (2018-present)
David A Sweetser, MD, PhD (2018-present)
Ronald Thibert, DO, MsPH (2018-present)
Holly I Welsh, MS; University of Missouri-Kansas City (2012-2018)
Lael Yonker, MD (2018-present)
Revision History
- 12 April 2018 (ha) Comprehensive update posted live
- 30 August 2012 (me) Review posted live
- 4 May 2012 (hha) Original submission
Publication Details
Author Information and Affiliations
MassGeneral Hospital for Children
Boston, Massachusetts
St Louis, Missouri
MassGeneral Hospital for Children
Boston, Massachusetts
MassGeneral Hospital for Children
Boston, Massachusetts
Massachusetts General Hospital
Boston, Massachusetts
Massachusetts General Hospital
Boston, Massachusetts
Publication History
Initial Posting: August 30, 2012; Last Update: April 12, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Sweetser DA, Elsharkawi I, Yonker L, et al. Pitt-Hopkins Syndrome. 2012 Aug 30 [Updated 2018 Apr 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.