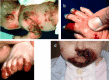
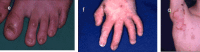
Molecular Pathogenesis
The proteins encoded by LAMA3, LAMB3, and LAMC2 assemble into the laminin 332 heterotrimer (aka LAM5 [Aumailley et al 2005]). A pathogenic variant in these genes can lead to reduced resistance to minor trauma and the resulting mucocutaneous blistering that is the hallmark of junctional epidermolysis bullosa (JEB) (reviewed by Kiritsi et al [2013]). The type of variant, the biochemical properties of the substituted amino acid, if present, and its location determine the severity of the blistering phenotype (see Genotype-Phenotype Correlations). Pathogenic nonsense variants predominate in the severe forms of JEB and result in the absence of one of the three proteins that assemble into laminin 332. Pathogenic missense variants in key positions of the protein subunits affect the ability of the laminin α3, β3, and γ2 polypeptides to assemble into a trimeric molecule, its secondary structure, and its ability to form the intracellular anchoring fibrils of the lamina densa.
Collagen XVII forms an integral part of the hemidesmosome and has an intracellular as well as extracellular component. There is evidence that it interacts with alpha-6 integrin within the hemidesmosome. The hemidesmosomes – structures made up of several protein components including COLXVII, alpha-6 beta-4 integrin, BPAG1, and plectin – anchor the epidermal cells to the underlying dermis. The type and position of variants in COL17A1 determine whether some partially functional protein is made and also affect the level of the cleavage plane of the skin. In some cases, variants affecting the intracellular domain result in a cleavage plane within the lowest level of the basal keratinocytes usually associated with epidermolysis bullosa simplex (EBS) [Charlesworth et al 2003].
Integrins associate in pairs containing one alpha and one beta chain. α6β4 integrin comprises one α6 and one β4 integrin from the integrin family of proteins and is a component of the hemidesmosomes of the epidermis. Integrins are known to participate in cell adhesion as well as cell-surface-mediated signaling. Insertion/deletion, splice junction, and amino acid substitution variants in both α6 and β4 integrin have been described and would result in EB-PA [Ruzzi et al 1997, Gache et al 1998, Lépinard et al 2000, Varki et al 2006, Masunaga et al 2015, Mencía et al 2016, Masunaga et al 2017].
COL17A1
Gene structure. The cDNA NM_000494.3 encodes 1,497 amino acids in 56 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Benign variants. There is one alternatively spliced mRNA variant [Ruzzi et al 2001].
Pathogenic variants. Pathogenic variants in COL17A1, which encodes the collagen XVII protein, a component of the hemidesmosome, typically result in JEB generalized intermediate [Gatalica et al 1997, Pulkkinen et al 1999, Takizawa et al 2000a, van Leusden et al 2001, Pasmooij et al 2004], although a few individuals with lethal JEB resulting from COL17A1 pathogenic variants have been described [Varki et al 2006, Murrell et al 2007]. All types of variants (including premature termination codon, nonsense, insertion/deletion, splice junction, and missense variants) distributed throughout the gene have been described. The type and location of the variants and the response of the cells to the variants determines the phenotype, which can range from mild to severe and in some cases lethal. Reversion to a normal phenotype has been described [Pasmooij et al 2005, Pasmooij et al 2012].
Normal gene product. Collagen XVII, NP_000485.3, (also known as BP180) has 1,497 amino acids and is composed of intracellular and extracellular domains separated by a transmembrane domain that distinguishes collagen XVII from other collagen family members. The intracellular domain is localized within the basal keratinocyte; the ectodomain is localized outside the cell and serves as an association point with other components of the basement membrane zone. The carboxy-terminal half of collagen XVII, a stretch of 916 amino acids, consists of 15 collagen domains of variable length (15 to 242 amino acids) that are separated by short stretches of non-collagen sequences. The collagenous domains associate to form a homotrimeric triple helical segment of the molecule characteristic of all collagen family members.
Abnormal gene product. Premature termination codon pathogenic variants that result in a null allele cause skin fragility, dental abnormalities, and alopecia usually found in individuals with JEB generalized intermediate. Other pathogenic variants may result in varying phenotypic severity. Although COL17A1 variants do not usually result in lethality, several cases of a neonatal lethal phenotype were described [Varki et al 2006, Murrell et al 2007]. Pathogenic variants that affect the intracellular domain may result in a cleavage plane more consistent with EBS and be misleading in terms of diagnosis based on electron microscopy biopsy results. Pathogenic variants that affect the transmembrane domain may result in intracellular accumulation of collagen XVII protein. Although glycine substitutions in COL17A1 have been described, no autosomal dominant variants resulting in skin fragility have been identified. Heterozygous carriers of a glycine substitution [Nakamura et al 2006] or other COL17A1 variants [Murrell et al 2007] may exhibit dental enamel pitting and this characteristic may be diagnostic for COL17A1 pathogenic variants in a family with an affected child [Murrell et al 2007].
ITGB4
Gene structure. The normal full-length cDNA is encoded in 41 exons spanning 36 kb of the genomic DNA. The cDNA comprises 6,033 bp with an open reading frame of 5,469 nucleotides encoding 1,822 amino acids. Two splicing variants express different isoforms of the protein [Pulkkinen et al 1997c]. The most common epidermal variant does not express exon 33. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. More than 100 pathogenic variants spanning all of ITGB4 have been described in individuals with EB-PA [Pulkkinen et al 1997b, Pulkkinen et al 1997c, Pulkkinen et al 1998a, Pulkkinen et al 1998b, Ashton et al 2001, Nakano et al 2001, Iacovacci et al 2003, Varki et al 2006, Masunaga et al 2015]. Pathogenic variants that cause premature termination codons on both alleles result in the most severe phenotypes, which are frequently lethal in the neonatal period. Other types of variants including amino acid substitutions and splicing variants may result in a less severe phenotype [Mellerio et al 1998, Pulkkinen et al 1998b, Chavanas et al 1999, Varki et al 2006]. In one individual severe blistering without pyloric atresia was described from a homozygous missense variant in ITGB4 [Inoue et al 2000] and in another a homozygote with missense and PTC ITGB4 variants [Inoue et al 2000, Jonkman et al 2000]. Few recurrent pathogenic variants have been described; however, the variant p.Cys61Tyr is common in individuals of Hispanic ancestry with JEB-PA in the US [Varki et al 2006]. See Table 3.
Table 3.
ITGB4 Pathogenic Variants Discussed in This GeneReview
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. Integrins associate in pairs containing one alpha and one beta chain. α6β4 integrin comprises one α6 and one β4 integrin protein from the integrin family of proteins and is a component of the hemidesmosomes of the epidermis. Within the hemidesmosome, α6β4 integrin forms attachments with collagen XVII to fulfill its role in the network of protein giving the epidermal strength and integrity and anchoring the epidermal cells to the underlying dermis through adhesion of the hemidesmosomes to the basement membrane. α6β4 integrin has also been shown to be involved in cell signaling and may play a role in carcinogenesis [Chung et al 2004, Guo et al 2006, Yoon et al 2006, Folgiero et al 2007].
Abnormal gene product. Null alleles may result in little or no protein seen with staining with anti-α6β4 integrin antibodies. Reduced staining was seen in some milder cases resulting from amino acid substitutions or splice junction variants.
LAMA3
Gene structure. All of LAMA3 is encoded in 76 exons spanning 318 kb on chromosome 18q11.2. Transcript variants are produced by alternative splicing (see Table 4 and McLean et al [2003]). For a detailed summary of gene information along with additional transcripts and protein isoforms, see Table A, Gene.
Pathogenic variants. Nonsense, missense, splicing, and insertion/deletion variants have been reported [Nakano et al 2002a, Varki et al 2006]. Premature termination codon pathogenic variants on both alleles result in JEB generalized severe in most instances. A few mildly affected individuals with JEB with premature termination codon variants have been reported [Nakano et al 2002a]. Amino acid substitutions and splicing variants may result in a milder phenotype [Posteraro et al 1998, Nakano et al 2002a]. Overlapping phenotypes in which pathogenic variants in LAMA3 result in skin fragility with eye and laryngeal involvement may exist [Varki et al 2006, Figueira et al 2007].
Table 4.
LAMA3 Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.151dupG 1 | p.Val51GlyfsTer4 |
NM_000227.4
NP_000218.3
|
| c.1981C>T 2 | p.Arg661Ter |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
- 2.
Cited by McGrath et al [1996] as c.1948C>T (p.Arg650Ter) without a reference sequence. Coordinates here from citation in OMIM 600805, allelic variant 0.0002 with dbSNP identification of rs17852757, which gives the above HGVS coordinates for the reference sequence in this table.
Table 5.
LAMA3 Transcript Variants and Protein Isoforms Discussed in This GeneReview
View in own window
| Transcript Variants | Protein Isoforms |
|---|
| Name(s) | Accession # | Length (nt) | Total # Exons | Name(s) | Accession # | Amino Acids (#) | Encoded by Exon #s 1 |
|---|
| 2, 3a |
NM_000227.4
| 5,623 | 38 | α3a, 2, LAMA3a |
NP_000218.3
| 1,724 | 39-76 |
| 1, 3b1 | NM_198129.2 2 | 10,666 | 75 | α3b1, 1, LAM3b1 |
NP_937762.2
| 3,333 | 1-38 & 40-76 |
| 3, 3b2 |
NM_001127717.2
| 10,498 | 74 | α3b2, 3, LAM3b2 |
NP_001121189.2
| 3,277 | 1-9, 11-38, & 40-76 |
- 1.
- 2.
Longest transcript; the genomic sequence of LAMA3 should be sequenced for diagnostic purposes (Table 1, footnote 8).
Normal gene product. There are three isoforms (α3a, α3b1, α3b2) produced by alternative splicing (Table 4). Note that the shorter α3b2 isoform of 1,724 amino acids is encoded in 38 exons (exons 39-76 of LAMA3) and is unique in that exon 39 is expressed.
The laminin A3 protein associates with laminin B3 and C2 proteins to form the laminin 332 heterotrimer that comprises the anchoring fibrils in the epidermis. The anchoring fibrils hold the layers of the basal lamina together and form associations with collagen VII on the dermal side and plectin and α6β4 integrin in the hemidesmosomes on the epidermal side. This interaction allows the formation of the protein network of the epidermis, which results in a flexible and resilient barrier to resist trauma.
Abnormal gene product. See Molecular Pathogenesis. In all three genes (LAMB3, LAMC2, and LAMA3), amino acid substitutions, splicing variants, and in-frame deletions and insertions may result in the formation of some partially functional protein that results in a milder phenotype. Specific amino acid substitutions, such as replacement of cysteine residues, inhibit the formation of disulfide bonds, result in altered laminin 332 intra- and intermolecular associations, and may result in a more severe phenotype. On a skin biopsy studied with immunofluorescence, if synthesis of one of the proteins is disrupted, the staining for the other two proteins will usually also be affected.
LAMB3
Gene structure. The normal LAMB3 cDNA has an open reading frame of 3,516 nucleotides in 23 exons spanning 29 kb. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Nonsense, missense, splicing, and insertion/deletion variants have been reported [Nakano et al 2002b, Varki et al 2006]. A few mildly affected persons with premature termination codon variants have been reported [Pulkkinen et al 1998a, Nakano et al 2002a]. Amino acid substitutions and splicing variants may result in a milder phenotype [Mellerio et al 1998, Posteraro et al 1998, Nakano et al 2002a]. The following LAMB3 pathogenic variants, p.Arg635Ter, p.Gln243Ter, c.957ins77, p.Arg42Ter are present in approximately 45% of individuals with H-JEB in the US [Varki et al 2006]. These pathogenic variants invariably result in premature termination codons and when found on both alleles result in JEB generalized severe.
Table 6.
LAMB3 Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.124C>T | p.Arg42Ter |
NM_000228.2
NP_000219.2
|
| c.727C>T | p.Gln243Ter |
| c.957ins77 | p.Glu320Ter |
| c.1903C>T | p.Arg635Ter |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. The laminin B3 protein has 1,172 amino acids. It associates with laminin A3 and C2 proteins to form the laminin 332 heterotrimer that comprises the anchoring fibrils in the epidermis.
Abnormal gene product. See Molecular Pathogenesis. In all three genes (LAMB3, LAMC2, and LAMA3), amino acid substitutions, splicing variants, and in-frame deletions and insertions may result in the formation of some partially functional protein that results in a milder phenotype. Specific amino acid substitutions, such as replacement of cysteine residues, inhibit the formation of disulfide bonds, result in altered laminin 332 intra- and intermolecular associations, and may result in a more severe phenotype. On a skin biopsy studied with immunofluorescence, if synthesis of one of the proteins is disrupted, the staining for the other two proteins will usually also be affected. Reversion by LAMB3 mosaicism to a normal phenotype has been described and has implications for treatment [Pasmooij et al 2007].
LAMC2
Gene structure. Two LAMC2 transcript variants result from alternative splicing. The longer LAMC2 cDNA transcript NM_005562.2 is expressed in the epidermis and encodes 1,193 amino acids in 23 exons spanning 55 kb. The shorter transcript variant, NM_018891.2, is expressed in the cerebral cortex, lung, and distal tubules of the kidney. For a detailed summary of gene, transcript, and protein information, see Table A, Gene.
Pathogenic variants. Nonsense, missense, splicing, and insertion/deletion variants have been reported [Castiglia et al 2001, Nakano et al 2002b, Varki et al 2006]. Amino acid substitutions and splicing variants may result in a milder phenotype [Posteraro et al 1998, Castiglia et al 2001, Nakano et al 2002a].
Table 7.
LAMC2 Pathogenic Variants Discussed in This GeneReview
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. The laminin C2 protein associates with laminins A3 and C2 to form the laminin 332 heterotrimer that makes up the anchoring fibrils in the epidermis.
Abnormal gene product. See Molecular Pathogenesis. In all three genes (LAMB3, LAMC2, and LAMA3), amino acid substitutions, splicing variants, and in-frame deletions and insertions may result in the formation of some partially functional protein that results in a milder phenotype. Specific amino acid substitutions, such as replacement of cysteine residues, inhibit the formation of disulfide bonds, result in altered laminin 332 intra- and intermolecular associations, and may result in a more severe phenotype. On a skin biopsy studied with immunofluorescence, if synthesis of one of the proteins is disrupted, the staining for the other two proteins will usually also be affected.