A large number of retroviruses infect cells that mediate the immune response, and a subset of these agents cripple the ability of the system to functional normally (Table 7). Although some of these effects were discovered almost 40 years ago (Laterjet and Duplan 1962; Anderson et al. 1971; Bendinelli et al. 1985), this topic received little attention until the devastating consequences of HIV infection became clear (see Chapter 11. The AIDS epidemic stimulated the hope that common pathogenic mechanisms would be involved in all retrovirus-induced immunodeficiencies and that simple animal models would be available for the study of HIV pathogenesis. Indeed, this prompted investigators to name the murine, feline, and nonhuman primate immunodeficiencies induced by simple retroviruses MAIDS, FAIDS, and SAIDS, respectively. Unfortunately, these disorders do not involve pathogenic mechanisms similar to those important in AIDS. Indeed, none of the diseases is related to each other mechanistically. Adding to the confusion, immunodeficiencies induced in cats and nonhuman primates by FIV and SIV are also sometimes referred to as feline and simian AIDS, respectively. Both of these syndromes share many features with AIDS (see Chapter 11, but they have little in common with the immunodeficiencies induced by simple retroviruses.
Table 7
Retroviruses That Induce Immunodeficiency Disorders.
Even though the pathogenesis of many of the retrovirus-induced immunodeficiencies is distinct from that of AIDS, study of them provides valuable insight into the mechanisms by which the immune response can be impaired. Indeed, the simple observation that retroviruses do not induce immunodeficiency by a common pathway is significant. The varied pathogenic mechanisms reflect the complex interactive network of specific and nonspecific cell-cell interactions required for normal immune function and the ability of a single type of immune cell to effect multiple responses (for review, see Janeway and Travers 1994). These features explain how the various viruses can act at many different points to elicit similar responses. These same properties and gaps in our understanding of the normal immune response complicate analyses of the pathogenesis of immunodeficiency. In addition, many of the features of the immune response that appear to be critical to pathogenesis in vivo cannot be studied in vitro, and well-defined virologic, genetic, and immunological tools are not available in all cases. The complexities of these types of studies can be illustrated by a discussion of MAIDS and FAIDS. For comparison, a discussion of FIV pathogenesis is also included. More detailed information on these and other viruses that induce immunodeficiency can be found in recent reviews (Arthur et al. 1986; Desrosiers and Letvin 1987; Wong 1990; Morse et al. 1992; Hoover and Mullins 1993; Pedersen 1993).
MAIDS
MAIDS was one of the first retrovirus-induced immunodeficiency disorders described (Latarjet and Duplan 1962; Legrand et al. 1981; Mosier et al. 1985). Although mice injected with a cocktail of viruses recovered from radiation-induced thymic lymphomas were first thought to develop a B-cell proliferative disorder, further study of these animals revealed that they were suffering from severe immunodeficiency. Mice with MAIDS display marked lymphadenopathy and splenomegaly resulting from proliferation of B cells, macrophages, and CD4+ T helper cells. Despite the dramatic increases in these cells, they do not respond to normal signals, and afflicted animals experience severe immunodepression (Mosier et al. 1985; Morse et al. 1989; Cerny et al. 1990a; Fitzpatrick et al. 1992). In the final stages of the disease, pulmonary function is impaired by lymphocytic infiltrates and enlarged thoracic lymph nodes, and the animal succumbs. Rarely, animals develop B- and T-cell lymphomas (Klinken et al. 1988; Chattopadhyay et al. 1991; Kubo et al. 1992; Tang et al. 1992; Huang et al. 1995; Simard et al. 1995). Very little is known about the T-cell tumors, but some of the B-cell tumors have new proviral insertions at the Spi1 locus (Huang et al. 1995) and probably arise via mechanisms that are similar to those involved in tumor induction by other retroviruses that lack oncogenes (see above Oncogenesis, Tumor Induction by Simple C-type Retroviruses That Lack Oncogenes).
Although both CD4+ T cells and B cells are required for development of MAIDS (Mosier et al. 1987; Yetter et al. 1988; Cerny et al. 1990b; Kim et al. 1994), cells of the B lineage are the primary target cell of the virus (Huang et al. 1989, 1991; Simard et al. 1994), and their growth is stimulated by viral infection. Infection stimulates growth of both the infected cells and neighboring uninfected B cells and CD4+ T cells by an unknown mechanism. Sustained proliferation seems to require interaction between B cells and CD4+ T cells and probably involves T helper cell functions analogous to those provided during a normal immune response (Giese et al. 1994). This interaction leads to polyclonal B-cell activation and hypergammaglobulinemia (Legrand et al. 1981; Mosier et al. 1985; Klinman and Morse 1989). As the disease progresses, the levels of immunoglobulin decrease and cells that resemble immature B cells accumulate (Hartley et al. 1989; Klinman and Morse 1989; Simard et al. 1994). The CD4+ cells also become refractory to stimulation, and most of them display a phenotype similar to that of memory and activated T cells (Morse et al. 1989; Holmes et al. 1990; Muralidhar et al. 1992). This picture is consistent with antigen-driven stimulation that leads to a nonresponsive state called anergy and suggests that an immune response is important in pathogenesis.
Virus Determinants Important for MAIDS
Analyses of the original mixtures of the Duplan virus (Latarjet and Duplan 1962) revealed that MAIDS induction requires a replication-defective virus that expresses an altered gag gene product (Fig. 18) (Aziz et al. 1989; Chattopadhyay et al. 1989). This protein, called Pr60 Gag, is myristoylated and localized to the inner surface of the plasma membrane like other Gag precursor proteins (Huang and Jolicoeur 1990, 1994; Hugin et al. 1991). However, the molecule contains substitutions in the carboxy-terminal region of the MA protein and several changes in p12, and it is not processed (Aziz et al. 1989; Chattopadhyay et al. 1991). Although helper virus appears to accelerate the onset of the disease (Chattopadyay et al. 1991), helper-virus-free stocks expressing only Pr60 Gag induce all of the changes associated with the disorder (Huang et al. 1989, 1991; Pozsgay et al. 1993b). The ability of a gag-encoded protein to induce immunodeficiency has not been observed in any other retrovirus-induced diseases nor do any of these other disorders arise in the absence of viral replication. These features highlight the unique nature of MAIDS pathogenesis.
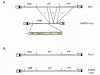
Pathogenesis of MAIDS Virus
Although it is apparent that expression of Pr60 Gag is necessary for induction of MAIDS, the mechanism by which immunodeficiency is induced is not well understood. The requirement for expression of MHC class II molecules and for the presence of CD4+ cells supports a role for the immune response in MAIDS development (Yetter et al. 1988; Giese et al. 1994). In addition, the accumulation of CD4+ cells that resemble memory and activated T cells argues that these cells have been stimulated by normal immune mechanisms (Holmes et al. 1990; Muralidhar et al. 1992). The nonresponsive character of these cells suggests that an eventual consequence of the response is T-cell anergy, a state in which T cells are no longer capable of responding when stimulated by antigen. B cells in MAIDS virus-infected mice are also abnormal and display a reduced capacity for normal signaling through the B-cell receptor (BCR) in response to antigen (Selvey et al. 1995).
The way in which MAIDS virus infection mediates immune breakdown remains a matter of some controversy. Initially, a superantigen response to a MAIDS virus-encoded protein appeared to be involved (Hugin et al. 1991; Kanagawa et al. 1992; Selvey et al. 1993). However, cells expressing only Pr60 Gag do not induce a superantigen response, even in vitro (Pozsgay et al. 1993a; Doyon et al. 1995), and other features that might be expected for a superantigen-driven disorder cannot be documented in MAIDS-infected mice (Fitzpatrick et al. 1992; Huang et al. 1992; Morse et al. 1992; Gilmore et al. 1993; Selvey et al. 1993; Koch et al. 1994; Doyon et al. 1995). Another model suggests that Pr60 Gag acts like an oncogene and stimulates uncontrolled B-cell proliferation, leading to immunodeficiency (Jolicoeur 1991; Jolicoeur et al. 1991b). However, this model does not explain the requirement for CD4+ T cells or the nonresponsive character of the expanded T cells. In addition, evidence that Pr60 Gag expression can alter B-cell growth directly has not been presented. The possibility that a broadly reactive antigen-driven response, perhaps occurring in the absence of costimulatory molecules, is important in establishing the disease also deserves consideration. Perhaps expression of costimulatory molecules that are required for normal regulation of the immune response is affected by Pr60 Gag.
FAIDS
FAIDS is a common immunodeficiency disorder that afflicts domestic cats. Indeed, this disorder is observed more frequently than malignant diseases (Hardy 1982; Hoover and Mullins 1993). Diseased cats show weight loss and intractable diarrhea. The numbers of B cells and both CD4+ and CD8+ T cells are decreased and those that remain are compromised in their ability to respond to mitogens or antigens (Hoover et al. 1987; for review, see Hoover and Mullins 1993). Unlike MAIDS, the B-cell defect appears to be secondary, reflecting the loss of T-cell function. In naturally infected cats, FAIDS is frequently accompanied by infections with other viruses and bacteria. This disorder appears to heighten the susceptibility of cats to infection with FIV, a lentivirus that also induces immunodeficiency (Pedersen et al. 1990; Beebe et al. 1994b; see below FIV-induced Immunodeficiency).
FAIDS Viral Determinants and Disease Induction
Viruses associated with FAIDS were isolated initially as complex mixtures of FeLVs from pet cats with immunodeficiency disease (for review, see Hoover and Mullins 1993). Cloning of one of these mixtures led to the isolation of a virus that induces FAIDS (Mullins et al. 1986; Overbaugh et al. 1988b). Although the original isolate had multiple mutations, sequences encoding the amino-terminal portion of SU and others encoding the carboxy-terminal portion of SU were linked to FAIDS induction (Fig. 18) (Quakenbush et al. 1990; Donahue et al. 1991; Overbaugh et al. 1992). As a consequence of these changes, the Env protein encoded by FAIDS virus is incapable of establishing superinfection resistance (Kristal et al. 1993; Reinhart et al. 1993). This defect allows the accumulation of large amounts of unintegrated proviral DNA in cells infected in vitro and in vivo and causes cell lysis (Mullins et al. 1986; Donahue et al. 1991). Thus, the primary cause of T-cell depletion in FAIDS appears to be direct lysis of infected T cells. Cell death mediated by accumulation of large amounts of unintegrated proviral DNA appears to be important in myeloblastosis-associated virus (MAV)-induced osteopetrosis and probably has a role in REV-induced wasting disorders and AIDS (see sections on Neurological Diseases and Other Retrovirus-induced Diseases and Chapter 11. The FAIDS model is particularly well-suited to investigations of this phenomenon and is one area in which studies of FAIDS could enhance our understanding of AIDS pathogenesis.
FIV-induced Immunodeficiency
The isolation of a lentivirus from pet cats with a severe immunodeficiency syndrome revealed that FeLV was not the only feline retrovirus capable of inducing immunodeficiency (Pedersen et al. 1987). This virus, named FIV, induces a disorder that shares many features with AIDS (see Chapter 11. Even though FIV appears to have a broader cell tropism in vivo, infecting both CD4+ and CD8+ T cells, macrophages, and B cells (Brunner and Pedersen 1989; Brown et al. 1991; English et al. 1993), the pathogenic mechanisms involved in both diseases are thought to be very similar. End-stage FIV-induced disease is marked by severe depression in the numbers of CD4+ T cells, opportunistic infections, and severe weight loss; sometimes, neurologic symptoms or tumors of lymphoid or myeloid cells are found (for review, see Pedersen and Barlough 1991; Pedersen 1993).
Clinical Stages of FIV-induced Immunodeficiency
FIV-induced immunodeficiency follows a course described by clinical stages patterned after those used for HIV (Ishida and Tomoda 1990). These include an initial, self-limiting acute disease in which the virus infects lymphoid cells in a pattern that mimics that observed in HIV infection (Barlough et al. 1991; Callanan et al. 1992; Bach et al. 1994; Beebe et al. 1994a; Dua et al. 1994; Diehl et al. 1995). This phase is followed by an asymptomatic period during which decreased numbers of CD4+ T cells and changes in the normal CD4+/CD8+ ratio are found (Ackley et al. 1990; Barlough et al. 1991; Torten et al. 1991; Walker et al. 1994). Decreased responsiveness of T cells to mitogen stimulation is also observed (Hara et al. 1990; Taniguchi et al. 1990, Siebelink et al. 1991). Following this phase, a period of generalized ill health is often documented, usually followed by a pre-AIDS-like disorder (see Chapter 11 (Ishida and Tomoda 1990; Shelton et al. 1990). Many diseased cats are euthanized at this stage.
The latent period for disease induction appears to be about 5 years. However, whether all infected cats develop immunodeficiency is not known. Tracking the natural course of infection is often difficult because most of the cats that have been studied are pets that were seen by veterinarians long after the initial infection. In one study, 4 of 11 FIV-infected, asymptomatic cats progressed to a stage similar to pre-AIDS during a 2-year observation period and two died (Ishida et al. 1992). About half of the specific pathogen-free cats that are inoculated with FIV and housed pathogen-free develop severe immunologic disorders in 2–4 years (Barlough et al. 1991; Torten et al. 1991).
Possible Cofactors
Many cats that are naturally infected with FIV are also infected with other feline viruses, especially FeLV (see above Oncogenesis, Common Molecular Themes in Oncogenesis) and feline syncytium-forming virus, a spumavirus that does not appear to cause disease (Pedersen 1987). Coinfection with FeLV may accelerate the development of FIV-induced disease (Grindem et al. 1989; Ishida et al. 1989; Pedersen et al. 1990), but direct interactions between FIV and FeLV probably do not occur in the coinfected animals (Beebe et al. 1994b). In addition, FeLV does not induce the lymphomas that are sometimes observed in end-stage FIV immunodeficiency; neither FeLV nor FIV genomes are detected in these cells (Terry et al. 1995). Because a very high percentage of FIV-infected cats are also infected with feline syncytium-forming virus (Yamamoto et al. 1989; Bandecchi et al. 1992), it is difficult to know if the presence of both of these viruses influences pathogenesis.
FIV Strains and Pathogenesis
FIV infection is common among domestic cats worldwide and, as with all retroviral infections, infected cats carry the virus for life (Yamamoto et al. 1988). Multiple isolates have now been studied and classified into four subtypes, A, B, C, and D, based on the sequence of the Env protein (Sodora et al. 1994; Kakinuma et al. 1995). Although viruses within a particular subtype are usually quite similar, considerable variation within env has been detected (Olmsted et al. 1989; Talbot et al. 1989; Phillips et al. 1990; Maki et al. 1992). The different subtypes are usually localized to particular geographic regions and reflect the population of virus circulating in cats in a particular area. These patterns of variation are quite similar to those observed among isolates of HIV (see Chapter 11. Unfortunately, very little is known about the viral determinants that are linked to pathogenicity.
Publication Details
Copyright
Publisher
Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY)
NLM Citation
Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997. Retrovirus-Induced Immunodeficiencies.
