Reviewed in this section are principles of immunology relevant to vaccine development and immunotherapeutics. Different vaccine and immunotherapeutic approaches are then discussed, with a comparison of the different kinds of immunological protection that can be expected. Finally, the experience with different retroviruses is discussed.
Immunological Principles Relevant to Vaccine and Immunotherapeutic Development
A brief overview is provided here, but for a more detailed discussion, see several excellent texts by Abbas et al. (1991), Paul (1993), and Janeway and Travers (1994).
Cells of the Immune System
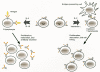
The immune system is made up of many (but not all) cells of the hematopoietic system. Immune system cells can be roughly divided into lymphocytes that specifically recognize foreign substances (antigens) and effector cells that attack foreign microbes or virus-infected cells upon stimulation by lymphocytes or lymphocyte products (Fig. 1). Lymphocytes can be divided into B cells, whose ultimate role is to secrete antibodies, and T cells. T lymphocytes specifically bind to “foreign” cells and are divided into two main types: helper (T helper) lymphocytes and cytotoxic T lymphocytes (CTLs). T-helper lymphocytes provide growth stimuli to both maturing B lymphocytes and CTLs (see below), whereas CTLs kill cells to which they bind (e.g., virus-infected cells). B lymphocytes provide the humoral arm of the immune system, and T lymphocytes make up its cellular arm.

Figure 1
Cells of the blood. The major cells of the blood are shown, and their major functions are indicated. (Modified from Fan et al. 1994.)
There are several different kinds of immune effector cells. Of major importance for viral infections are phagocytes and natural killer (NK) cells. The two types of phagocytes are the neutrophils and macrophages (whose circulating forms are called monocytes). Neutrophils engulf and destroy bacteria and macrophages destroy virus-infected cells. NK cells bind to and destroy virus-infected cells and some tumor cells.
Humoral Immunity
Antibodies
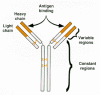
Antibodies are the key components of the humoral immune system. An antibody molecule is a tetrameric protein consisting of two identical heavy chains and two identical light chains (Fig. 2). Both heavy and light chains contain variable and constant regions, i.e., regions that vary among different antibodies and regions that are shared. Each antibody molecule binds a specific antigen, or epitope; binding specificity is dictated by the primary sequence of the variable regions of the heavy and light chains, which in turn dictates the overall structure of the variable regions. The constant regions carry out other functions, including antibody oligomerization, determining whether the molecule is localized in the membrane or is secreted, and signaling of effector cells.
Generation of a Humoral Immune Response
Two general principles underlie the generation of a humoral immune response: (1) Each B lymphocyte makes antibodies with only one antigenic specificity and (2) exposure to an antigen leads to clonal selection and maturation of preexisting B lymphocytes that produce antibodies specific for that specific antigen.
B lymphocytes are continually developing in an individual (see also Chapter 10. During the differentiation process, each developing B lymphocyte undergoes DNA rearrangements that produce the functional genes encoding the immunoglobulin heavy and light chains. For the light chain, these DNA rearrangements randomly combine one of several possible V-region genes with one of several J-region genes and with a constant region gene (Fig. 3). For the heavy chain, a similar process takes place, except that the variable region results from random combinations of V, D, and J gene segments. During recombination, two other molecular events occur that contribute to diversity: random addition of nucleotides at V-D and D-J junctions during heavy-chain VDJ joining (N-region or junctional diversity), and high-frequency base substitutions in the variable regions (somatic mutations). These events collaborate to produce, from a fixed number of germ-line immunoglobulin structural genes, a population of B lymphocytes that make antibodies which recognize virtually any antigen. (Although the mechanism described here pertains to many species, including humans and mice, other species, e.g., chickens, use somewhat different molecular mechanisms to generate antibody diversity.)
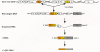
During maturation of B lymphocytes, a second kind of DNA arrangement occurs: class switching for the heavy-chain constant regions. The initial VDJ recombination for the heavy-chain genes joins the variable region segments to the μ constant region (Fig. 4). This leads to production of antibodies of the IgM class. During maturation of the B lymphocyte, “switch” recombination events occur that eliminate the appended μ constant region gene, bringing the rearranged VDJ region sequentially into proximity with other heavy-chain constant regions (e.g., γ, α, and δ). This leads to production of antibodies of different classes (e.g., IgG, IgA, IgD, and IgE) in the progeny lymphocytes, all with the same VDJ specificity. Antibodies of different classes have different properties, for example, the ability to signal effector cells. For instance, antibodies of the IgG class are found in the circulation, antibodies of the IgA class are secreted in mucosal tissues (mucosal immunity is discussed below), and antibodies of the IgE class signal attack by mast cells.

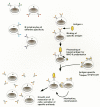
Cellular events involved in development of a primary immune response are illustrated in Figure 5. Prior to encounter with antigen, immature B lymphocytes express antibodies on their cell surfaces in a membrane-bound IgM form. When these immature lymphocytes encounter a particular antigen, the antigen binds to those lymphocytes that produce an antibody specific for that antigen. This binding provides an activation signal to the lymphocytes that recognize the antigen. For most antigens and cognate B lymphocytes, “help” from a T-helper cell is also required. T lymphocytes produce antigen-specific surface molecules analogous to immunoglobulins (T-cell receptors are described below). Interaction of an antigen-specific T-helper lymphocyte simultaneously with the antigen and the B lymphocyte provides “help” to the B lymphocyte that is mediated by cytokines such as interleukin-4 and interleukin-10 (IL-4 and IL-10). T-cell help results in the division and maturation of B cells, including heavy-chain class switching. The net result is the production of large numbers of B lymphocytes that secrete antibodies specific for the encountered antigen. Mature B lymphocytes are called plasma cells.
The response resulting from the first encounter with an antigen is called the primary immune response. In experimental systems in which animals are treated with strong antigens in combination with potentiating compounds (adjuvants), circulating antigen-specific IgM antibodies can be detected in as little as 10–14 days. With time, the antibody titer rises, and other classes (IgG, IgA) appear. In other cases, the primary immune response may take much longer. For example, seroconversion to antibody positivity after HIV infection can take 6 months or more.
If the antigen is cleared by the antibodies of a primary immune response, production of specific antibody ceases and circulating concentrations fall to low or undetectable levels. However, dormant “memory” B cells that have undergone all of the maturation steps are still present. These dormant cells do not secrete antibody. A second encounter with the same antigen results in a secondary immune response, in which the memory B cells are mobilized rapidly. A secondary response is characterized by a more rapid appearance of circulating antibodies, the immediate appearance of immunoglobulin classes that are effective in eliminating the antigen (e.g., IgG and IgA), and prolonged production of specific antibody. This property of immune recall is the reason that some vaccines are given as multiple injections and for the periodic administration of “booster” inoculations.
It is important to point out that only a fraction of virus-specific antibodies can inactivate the virus. Such antibodies are called neutralizing antibodies. Many (and for some viruses most) antibodies that react with the virus do not have neutralizing activity, either because they recognize antigens (such as Gag and Pol proteins) that are not present on the viral surface or because their binding (e.g., to Env) is not sufficient to block viral binding or entry.
Mechanisms of Antibody Inhibition of Viral Infection
Virus-specific antibodies inhibit infection by several mechanisms. The most straightforward is direct inhibition by the antibody. Virus-specific antibodies can inactivate a virus by causing conformational changes in virion proteins, they can interfere with virus binding to the cell receptor, or they can interfere with early postbinding events (e.g., penetration and uncoating). These activities can be detected by incubation of the antibody with the virus prior to infection or by inhibition of viral infection in vitro.
A second mechanism for antibody inactivation is through the complement system, a system of circulating cellular proteins. Binding of one of the complement components to the constant (Fc) region of certain antibody classes (e.g., IgGs) when the antibodies are bound to infected cells (or enveloped viruses) leads to a cascade of proteolytic reactions involving other complement components that ultimately results in disruption of the membrane of the virus (or virus-infected cell).
Another immune mechanism that involves antibodies is antibody-dependent cellular cytotoxicity (ADCC). Certain immune effector cells (macrophages and NK cells) have receptors for the Fc portion of antibodies (IgGs). These effector cells can bind to a virus-infected cell that has antiviral antibodies bound to it (cells with antibodies bound to them are said to be “opsonized”) and eliminate the opsonized cells either by phagocytosis (macrophages) or by cell killing (NK cells). Similarly, mast cells bind to the constant regions of IgE molecules (including IgEs that are bound to virus-infected cells) and release reactive oxygen species that kill cells in the vicinity.
Antibody Enhancement
In some cases, antibodies can actually potentiate viral infection or exacerbate pathology. Several mechanisms for this potentiation have been documented. One well-known example involves dengue virus: The first time an individual is infected, the symptoms are relatively mild; however, a second dengue infection can result in dengue shock syndrome, which is mediated by the dengue-specific antibodies produced in response to the first infection (Halstead 1988). In retroviruses, antibodies potentiate the pathology of EIAV infections. The anemia associated with the acute phases of EIAV infection is believed to result from antibody-mediated destruction of red blood cells that passively bind viral glycoprotein (Montelaro et al. 1989). Finally, in vitro enhancement of HIV and SIV (simian immunodeficiency virus) infection by antibodies from infected individuals has been reported (see below). It may be advisable to eliminate Env sequences that induce enhancing antibodies from candidate vaccines.
Mucosal Immunity
Another important issue is mucosal immunity. Antibodies not only circulate in the blood, but they are also present in secretions of the intestinal tract, the respiratory tract, the oropharynx, and the female reproductive tract. These antibodies are typically of the IgA class and are produced by lymph nodes associated with those tissues (e.g., the gut-associated lymphoid tissues). For many viruses that encounter the host through these tissues (e.g., respiratory, enteric, and venereally transmitted viruses), mucosal immunity may be of greater importance for protection from infection than the circulating antibodies. Procedures that efficiently induce mucosal and circulating immunity are important in vaccine design.
Cell-mediated Immunity
The T-cell Antigen Receptor
T lymphocytes have on their surfaces T-cell antigen receptor molecules that are analogous to immunoglobulins. Like immunoglobulins, T-cell receptors (TCRs) are tetramers of large (α) and small (β) chains with variable and constant regions generated by processes analogous to those that are used by B lymphocytes to produce immunoglobulin (generation of diversity by gene rearrangement and clonal selection). However, class switching does not occur for T lymphocytes, and the TCR molecules remain embedded in the cell surface. Thus, antigenic recognition resides in the T lymphocytes.
The Antigen Receptor Complex on T Lymphocytes
Unlike surface immunoglobulin in immature B lymphocytes, the TCR molecule is closely associated with several other cellular proteins on the surface of the T lymphocyte (Fig. 6). These include proteins of the CD3 complex and, depending on the cell type, either CD4 or CD8. CD4 and CD8 are proteins of the immunoglobulin superfamily (a family of genes that produce protein whose sequence and structure are related to those of immunoglobulins), and they have important cooperating roles in the recognition of antigen by T lymphocytes (see below).
Antigen Recognition and MHC Restriction
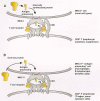
T lymphocytes differ from B lymphocytes and antibodies in that they specifically bind to antigens only when the antigens are held (or presented) on other cells by MHC (major histocompatibility complex) molecules. MHC molecules are also proteins of the immunoglobulin superfamily, and there are two types, class I and class II. MHC molecules are highly polymorphic: There are multiple genes for class I MHC and class II MHC proteins in the MHC complex, and multiple alleles for each gene. The MHC proteins hold small proteolytically processed portions of antigen (typically peptides 9–11 amino acids in length), and it is these peptides, in combination with a specific MHC molecule, that are recognized by the variable regions of the TCR in the antigen receptor complex (Fig. 7). Different MHC proteins have specific affinities for different antigenic peptides. As a result, a given peptide produced from a viral protein may be an efficient T-cell antigen in individuals who express a particular set of MHCs, whereas the same viral peptide may not be antigenic in individuals with different MHCs.

There are two requirements for T-cell recognition of an antigen on another cell. The first is self-MHC; i.e., a T lymphocyte will only interact with another cell if that other cell's MHC molecules are the same (or overlap) as those of the T lymphocyte. This results from selection for such T lymphocytes during their maturation in the thymus. With regard to natural infections, this does not present a problem since all cells in an individual (both T lymphocytes and infected cells) express the same MHC proteins. However, the design of vaccines intended to induce cell-mediated immunity must take this into account.
The second requirement for T-cell recognition is determined by whether the T lymphocyte has CD4 or CD8 protein in its antigen receptor complex. CD4+ lymphocytes only bind to cells that express MHC class II protein, and CD8+ lymphocytes only bind to cells that express MHC class I protein (Fig. 7). MHC class II protein is expressed predominantly by B lymphocytes, macrophages, and dendritic cells, whereas MHC class I protein is expressed by most other cell types. Thus, CD4+ lymphocytes will bind to B lymphocytes, dendritic cells, and macrophages if they contain the cognate antigen, and CD8+ lymphocytes will bind to other cells if they express the cognate antigen.
MHC class-I- and class-II-positive cells also differ in the way in which they process antigens. Class-II-positive cells (“antigen-presenting cells”) can take up antigens from outside by endocytosis, degrade them into small peptides, and reexport the peptides (now bound to MHC class II protein) to the cell surface (Harding and Unanue 1990). These peptide–MHC class II complexes can then be recognized by specific CD4+ lymphocytes. In contrast, MHC class I protein can, in general, form complexes with processed antigen peptides only if the antigen is synthesized in the same cell. As a consequence, CD8+ T lymphocytes can only bind to class-I-positive cells that are synthesizing the antigen (e.g., virus-infected cells).
T-helper Lymphocytes versus CTLs
As discussed above, T lymphocytes can be divided into two functional groups: T-helper lymphocytes and CTLs, based on whether they provide “help” to B and T lymphocytes or bind to and kill target cells. T-helper lymphocytes are exclusively CD4+ and most CTLs are CD8+, although there are some CD4+ CTLs as well. As a result, T-helper lymphocytes interact with MHC class-II-positive cells, whereas CTLs predominantly interact with MHC class-I-positive cells (Fig. 7).
The role of T-helper lymphocytes in humoral and cellular immunity is illustrated in Figure 8. T-helper lymphocytes provide growth stimuli to primed B lymphocytes and CTLs by secreting cytokines. For B lymphocytes, the stimulatory cytokines are IL-4 and IL-10, whereas for CTLs, IL-2 and interferon-γ (IFN-γ) are important. Recent evidence indicates that T-helper lymphocytes can be subdivided into two groups: TH1 cells produce IL-2 and IFN-γ in response to antigenic stimulation (and thus provide help to CTLs), and TH2 cells produce IL-4 and IL-10 (and thus help B lymphocytes) (Coffman et al. 1991). Moreover, there is the suggestion that TH1 cytokines may inhibit the growth of TH2 cells and vice versa.
Vaccine Approaches
Many different strategies are now being explored in an attempt to develop protocols that will provide an effective immunological intervention in retroviral infection (see Table 1). Traditionally, the goal of a vaccine has been to produce protective immunity prior to viral exposure. However, for retroviral infections (particularly HIV), vaccines or immunotherapeutics that can stimulate immunity in an already-infected individual may also be important. It should also be noted that most successful vaccines in use today do not completely eliminate or prevent infection, but they do reduce the level of the virus in the infected individual and as a consequence can eliminate the resulting disease.
Table 1
Vaccine Approaches to Retroviral Infection.
Passive Immunotherapy
This approach involves passage of antibodies or immune cells from an individual who has encountered an antigen or infectious agent into a naive individual. Although passive immunotherapy produces only short-term immunity, it has been used to treat acute infections.
Antiviral Antibodies
Antibodies (or inactivated serum) from individuals who are immune to an infectious agent can provide short-term protection. For instance, pooled γ-globulin is administered prophylactically to individuals who have been exposed to hepatitis A or B viruses (HAV, HBV). In principle, if strong neutralizing monoclonal antibodies to invariant viral epitopes can be developed, they might also be useful either prophylactically or therapeutically.
CTL Transfer
A major theoretical problem limits the usefulness of passive transfer of antiviral CTLs: the requirement for self-MHC for antigen recognition. Although adoptive transfer experiments in inbred mice readily demonstrate transfer of cellular immunity from immune to naive mice, the high MHC polymorphism in most outbred species (including humans) makes this strategy impractical.
Vaccines That Induce Humoral Immunity
Many of the vaccines now in use induce humoral immunity. The goal is the generation of neutralizing antibodies, with the final assessment of efficacy being disease prevention.
Inactivated Virus
This is one of the oldest methods of vaccination development, dating back to the Pasteur rabies vaccine. In practice, inactivated viral vaccines have been quite efficient in inducing protective immunity, including immunity to some nonprimate retroviruses. The efficacy of these vaccines may be related to the fact that the viral proteins are in a relatively native configuration. Moreover, this type of vaccination can produce neutralizing antibodies directed against multiple viral proteins and epitopes. One concern with inactivated viral vaccines is the danger of incomplete inactivation, which has, in the past, resulted in accidental infection of patients (e.g., in some early poliovirus vaccine trials).
Subunit Vaccines
These vaccines are based on one viral protein—typically an external virion protein, since these proteins are generally the targets for neutralization. The anti-HIV vaccines currently in clinical trials are mostly subunit vaccines based on either gp120 (SU) or gp160 (the precursor for the SU and TM [transmembrane] proteins). Subunit vaccines have the advantages that they are quite safe (complete viral particles are not present), and they can be generated by recombinant DNA methodologies. Disadvantages include the fact that such vaccines generally have been less efficient at inducing protective immunity. To some extent, this may reflect the fact that harsh chemical procedures are sometimes required to isolate and purify proteins produced by recombinant DNA techniques, which may affect their antigenic properties. It is also possible that antibodies against multiple viral proteins may be important in establishing effective protective immunity.
Peptide Vaccines
These vaccines consist of immunogenic subfragments of viral proteins, which can be either expressed through recombinant DNA techniques or synthesized chemically. In theory, peptide vaccines can include peptide sequences corresponding to multiple antigenic sites present in one or more virion proteins, thereby increasing the likelihood of an effective antibody response. Such a hybrid vaccine would be expected to stimulate T-helper lymphocytes specific for the hybrid sequences (see Fig. 5). For example, a bivalent peptide vaccine containing a cell determinant for the hepatitis B pre-S region fused to a T-helper determinant from the HBV core protein raises effective humoral responses (Milich et al. 1988). A synthetic peptide immunogen consisting of B-cell and T-helper determinants from HIV-1 SU protein has also been shown to induce neutralizing antibodies in goats (Palker et al. 1989).
Vaccines That Induce Cell-mediated Immunity as well as Humoral Immunity
Cell-mediated immunity and virus-specific CTLs can be as important as neutralizing antibodies in controlling viral infections. This has been demonstrated for several viruses (Ahmed et al. 1984; Salvato et al. 1991), including some retroviral infections (see below). Indeed, once infection has been established in an individual, virus-specific CTLs are likely to be quite important in eliminating virus-infected cells. This is particularly relevant for retroviruses because infection by these viruses does not usually cause cell death. As described above, CTLs are largely CD8+ and therefore interact predominantly with antigen-containing MHC class-I-positive cells. Since MHC class I antigen processing requires that the antigen be synthesized within the MHC class-I-positive cell, vaccines consisting of inactivated viruses or purified proteins are not likely to induce strong CTL responses, although CD4+ CTLs can develop. For this reason, the most effective vaccine approaches for stimulating cellular (as well as humoral) immunity will be those in which the target virus or viral proteins are expressed in the vaccinee.
In contrast to neutralizing antibodies largely directed against the external proteins of the virion, CTL responses may be directed against both internal and external virion proteins, as well as nonstructural proteins. This results from the fact that peptides from all of these proteins can be processed and presented on the surface of the infected cell by MHC molecules. This is a potential advantage in retroviruses in that there is considerably less variation for gag and pol in comparison to the env gene (the amino-terminal and central regions of reverse transcriptase are particularly well conserved); CTL-inducing vaccines directed against those proteins might be able to protect against more viral strains than those directed against env.
Attenuated Viruses
The best and most efficient method for establishing both cellular and humoral immunity is to use attenuated viruses. These viruses establish infection in the vaccinee, but they do not cause disease, e.g., the Sabin attenuated poliovirus vaccine. Vaccination with attenuated viruses has been shown to prevent infection and/or disease in animal retroviruses (see below). However, the possibility of mutation back to virulence, which has been documented for the attenuated poliovirus vaccine, is a major concern in the development of attenuated vaccines for human retroviruses, particularly given the long latencies and high mutation rates seen with these viruses.
Vaccinia-based Vaccines
Vaccinia virus (the virus used as a live vaccine for smallpox) has been modified by recombinant DNA techniques such that it can carry and express foreign proteins (Mackett et al. 1982; Piccini et al. 1987). Vaccinia recombinants that express proteins from other viruses have been developed as potential vaccines. Vaccinia has the advantages that it possesses a wide host range and that it is approved for use in humans. Vaccinia recombinants have been shown to induce CTL responses for vectored viral genes (see below). Potential difficulties exist with the use of recombinant vaccinia vaccines in humans. First, many individuals have already been immunized with vaccinia and are expected to be infected only inefficiently with a recombinant vaccine. Second, use of vaccinia recombinants as immunotherapeutics in HIV-infected individuals may be risky, since they are susceptible to serious systemic vaccinia infections (Mortality and Morbidity Weekly Report 40, No. RR-14). An additional question is whether it is advisable to stimulate T cells (by vaccinia infection) in HIV-1-infected individuals, since this could increase replication of HIV-1.
Some of the problems with vaccinia recombinants can be circumvented by using canarypox recombinants (Taylor et al. 1991). Canarypox is an avian poxvirus related to vaccinia, and similar recombinants carrying foreign genes have been generated. In contrast to vaccinia, canarypox infection in nonavian species results only in a limited initial infection. Although viral proteins are expressed, assembly of infectious virions does not occur, which should eliminate a systemic spread of infection. Moreover, potential human recipients have not been previously infected with canarypox, so preexisting immunity is not an issue.
Retroviral Vectors
Another method under development for generating CTL responses is retroviral vectors (see Chapter 9. Retroviral vectors engineered to express SIV and HIV proteins have been shown to induce CTL responses in animal models. Areas of concern include potential contamination with replication-competent retroviruses and achieving vector titers high enough to produce a strong immune response.
Direct DNA Injection
It has recently been shown that intramuscular injection of DNA can lead to uptake and expression of the DNA as protein (Wolff et al. 1990). This procedure can induce both humoral and cellular immunity, and it has been shown to protect mice from influenza virus infection (Fynan et al. 1993; Ulmer et al. 1993).
Current Experience with Retroviral Vaccines
Discussed in this section are the neutralizing antibody and cellular immune responses for each retroviral group if it is known. Identification of neutralizing, CTL, and T-helper epitopes on the different viral proteins may be important in designing vaccines. In addition, it is important to distinguish immunological responses specific for a particular viral strain (type-specific) from those that are reactive against multiple isolates (group-specific). In general, group-specific responses are the most important in vaccine design. Current experiences with different vaccine approaches are then described.
Murine Leukemia Viruses
Murine leukemia virus (MLV) is the best studied animal retrovirus. Studies on immune responses to MLV infection have been facilitated by the extensive knowledge of mouse genetics and the availability of immunological and molecular probes for the immune system of the mouse. Some of the commonly studied MLVs include Moloney MLV (Mo-MLV, which induces T lymphoma), Friend MLV (Fr-MLV, which induces erythroid leukemia), and the Friend MLV complex (which contains a replication-defective acute leukemia virus as well as a replication-competent Fr-MLV helper). In addition, the AKR strain of inbred mice spontaneously activates an endogenous virus (Akv MLV) that is leukemogenic in these mice.
Immune Responses to MLV Infection
The host's immune response to MLV infection is dependent on the infection protocol, the virus used, and the genetic background of the mouse. The standard method for infecting mice with retroviruses is inoculation of newborns, since this is the most efficient means of inducing leukemia. This is analogous to the spontaneous production of leukemia in AKR mice; these mice spontaneously express endogenous Akv MLV shortly after birth (see Chapter 10. Mice infected at birth by MLVs generally show little evidence of antiviral humoral or cellular immunity (Chieco-Bianchi et al. 1988). The lack of cellular immunity is probably the result of tolerization: The immune system in a newborn mouse is immature and infection at that time leads to recognition of MLV as “self” and virus-specific T lymphocytes are eliminated. Circulating anti-MLV antibodies are not produced in mice infected as newborns, but virus-antibody complexes have been detected in the kidneys in some cases (Oldstone et al. 1972; Chieco-Bianchi et al. 1988). Whether these antibodies have virus-neutralizing activity has not been determined.
If mice are infected as newborns with strains of MLV that replicate poorly, virus-specific CTL responses can be detected. Presumably, the relatively low levels of virus present soon after birth are insufficient to induce tolerance, but they are capable of inducing virus-specific CTLs. One example is AKR.Fv1 b mice (AKR mice congenic at the Fv1 b allele). The endogenous N-tropic Akv MLV does not replicate efficiently in these mice after it is activated, and the level of virus in these animals is low. In the appropriate genetic background, virus-specific CTLs can be detected (Green 1984). The CTLs are directed against the viral SU protein, and the CTL epitope has been mapped (Azuma et al. 1987; White et al. 1994). In this regard, it should be noted that CTL epitopes will be dependent on the MHC genotype, since different MHC class I and class II proteins show specific sequence preferences for the oligopeptides that they bind and present. This specificity is an important consideration in designing CTL-inducing vaccines. In another example, a long terminal repeat (LTR) variant of Mo-MLV (Delta Mo+SV Mo-MLV; Hanecak et al. 1988) that establishes low levels of infection in vivo induces a virus-specific CTL response in BALB/c mice, and the ability of this virus to induce a CTL response correlates with a long disease latency (E. Suchman and H. Fan, unpubl.).
Infection of adult mice with MLVs results in stronger antiviral immune responses that can influence the efficiency of infection and/or pathogenesis. Infection of C57BL/6 mice by Mo-MLV does not result in long-term viral infection or leukemia. However, pretreatment of the mice with antibody to CD4 (and elimination of CD4+ lymphocytes) prior to infection leads to establishment of viral infection and the development of leukemia (Biasi et al. 1991). This result implies that T-helper cells are necessary for suppressing viral replication and leukemogenesis in this system.
Unlike most MLVs, which are not pathogenic in adult animals, infection of adult mice with the Friend virus complex (FV; a mixture of Fr-MLV and the defective spleen focus-forming virus; see Chapter 10 causes erythroleukemia in some mouse strains, although infection of other strains does not. This system has permitted genetic mapping of determinants of resistance and sensitivity, and several have been shown to involve the host immune response to infection (Chesebro et al. 1990). Two resistance genes (rFv1 and rFv2), which map to the mouse MHC (H-2) region (Chesebro and Wehrly 1978; Chesebro et al. 1990), affect the cellular immune response to FV. The third gene (rFv3) does not map to H-2, but it does affect the production of virus-specific neutralizing antibody (Chesebro and Wehrly 1979; Chesebro et al. 1990).
Host immune responses have been studied in sensitive and resistant adult FV-infected mice; both neutralizing antibodies and CTL responses have been detected. Of the two, virus-specific CTLs appear to be crucial: Strong CTL responses are correlated with resistance to disease (Robertson et al. 1992). On the other hand, the presence of neutralizing antibodies (in rFv3 r strains) decreases levels of viremia, but erythroleukemia ultimately develops (Chesebro and Wehrly 1979). CD8+ CTL responses are directed against sequences in Env, whereas CD4+ responses are directed against Gag (Klarnet et al. 1989).
Vaccine Control of MLV Infection
Several different vaccine approaches intended to prevent MLV infection have been tried. Injection of a polyclonal anti-MLV env antibody can prevent infection by the FV complex (Schafer et al. 1976). This approach was also used to inhibit leukemia in AKR mice (Huebner et al. 1976; Schwarz et al. 1979; Weinhold et al. 1984). Interestingly, it was important to administer the antibody within the first few days after birth (Weinhold et al. 1984). Even under these conditions, infection by AKR MLV was not prevented, but the early administration of antibody apparently protected certain bone marrow cells from infection (Buckheit et al. 1988a,b). These experiments emphasize the critical difference between preventing infection in general and preventing infection of cells crucial for the development of disease.
Inactivated viruses and viral subunits have also been tested as vaccines for MLVs. An aggregated form of Fr-MLV gp70 (SU) protein was able to protect against disease following FV challenge (Kleiser et al. 1986), and inactivated Fr-MLV was found to be an efficient vaccine against Fr-MLV-induced disease (Chesebro 1990).
Attenuated MLVs have been shown to be effective vaccines. An N-tropic, nonpathogenic FV (FV-N) was found to confer effective protection against wild-type FV challenge in Fv1 b mice (Chesebro 1990). The vaccinated mice showed a strong virus-specific CTL response and produced neutralizing antibodies. There was a good correlation of protection and the CTL response, and resistance could be adoptively transferred to other mice by CD8+ cells, demonstrating the importance of an antiviral CTL response in protection against FV infection. It was noted in these studies that although vaccination reduced the viral burden significantly, low levels of FV-infected cells could still be detected in the challenged mice. Other attenuated Fr-MLV vaccines have been developed. In one case, a deletion in the env gene renders the virus replication-defective, but the defective virus can be produced by a retroviral packaging system (Ruan and Lilly 1992; see Chapter 9. This defective Fr-MLV prevented erythroleukemia when the mice were challenged with FV, and both antibody and CTL responses to Fr-MLV were detected.
An attenuated replication-competent Fr-MLV was found to prevent disease by subsequent challenge with a pathogenic strain, but in this case, protection appeared to be the result of viral interference (Corbin and Sitbon 1993). Viral interference can protect against MLV infection in mouse stains expressing endogenous MLV SU proteins. The Rmcf R gene confers resistance to infection by mink cell focus-inducing (MCF) recombinant viruses (see Chapter 10, and the Fv4 gene confers resistance to ecotropic MLV. For both of these genes, the mechanism of resistance has been shown to be expression of an endogenous MLV in the resistant cells (Ruscetti et al. 1985; Buller et al. 1988; Ikeda and Sugimura 1989).
A vaccinia recombinant expressing the Fr-MLV env gene has also been tested as an anti-FV vaccine (Chesebro 1990). This vaccine was less effective in protecting mice against FV challenge than either inactived whole FV or attenuated FV-N. Moreover, it did not induce neutralizing antibodies or CTLs prior to FV challenge.
Avian Retroviruses
Vaccine studies have not been carried out on avian retroviruses. However, there is an interesting effect of endogenous retroviral expression on exogenous viral infectivity and tumorigenesis. Chickens that carry active endogenous viruses (the ev loci; see Chapter 8 appear to develop lower levels of virus-specific neutralizing antibodies and to develop leukemia more rapidly after infection with avian leukemia virus (ALV), when compared to chickens that lack any endogenous ALVs (line 0) (Crittenden et al. 1984; Smith and Fadly 1988). These results suggest that fetal or neonatal expression of endogenous viral proteins causes at least a partial immunological tolerance to exogenous avian retroviruses. A defective endogenous virus (ev-6) that only expresses Env glycoprotein demonstrates this effect (Smith et al. 1991). In support of this hypothesis, infection of embryos with the nonpathogenic endogenous virus Rous-associated virus (RAV-0) in ovo can also increase the efficiency of infection and leukemogenicity of subsequent infection by exogenous RAV-1 or RAV-2 (Crittenden et al. 1987). These effects may well be operative for other animal species that carry and express endogenous retroviral sequences immunologically related to the exogenous viral pathogens that they acquire. On the other hand, chickens that express the subgroup A virus envelope from an experimentally introduced endogenous virus are resistant to infection by ALV of the same subgroup—presumably due to receptor interference (Crittenden and Salter 1992). ev expression can also protect against immunemediated ALV wasting disease.
Immunity to Rous sarcoma virus (RSV)-induced sarcomas has also been studied. Certain chicken lines are more resistant to the induction of tumors by RSV, and this resistance is associated with particular MHC alleles (Taylor et al. 1992). Thus, cell-mediated immunity is likely to be important for resistance to RSV-induced sarcomas. Indeed, bursectomy (removal of the bursa of Fabricius, leading to elimination of B lymphocytes) does not affect the susceptibility of resistant chickens to RSV tumorigenesis (Halpern et al. 1985). Expression of endogenous viruses also affects the immune response to RSV-induced tumors in a type-specific manner (McMahon et al. 1986).
Feline Leukemia Virus
Research on feline leukemia virus (FeLV) vaccines has been spurred by the fact that FeLV-induced lymphoma is a significant veterinary problem. Moreover, the fact that cats are outbred presents practical problems in vaccine development analogous to some of the problems encountered in attempts to produce vaccines for human retroviruses. Several commercial FeLV vaccines are now available, and the issues that arise in testing their relative efficacies are instructive.
Attempts to develop a prophylactic FeLV vaccine have been under way since the mid 1970s. Olsen and colleagues developed a vaccine consisting of proteins (predominantly Env) shed from an FeLV-infected lymphoid T-cell line that induced significant protection to challenge with FeLV (Lewis et al. 1981). This vaccine was marketed commercially as “Leukocell” beginning in 1985 (Pollock and Haffer 1991). The efficacy of this vaccine was studied in both laboratory settings and field trials. Although some studies reported significant although not complete protection from challenge infection (e.g., threefold reduction in risk; Haffer et al. 1987; Pollock and Scarlett 1990), equivalent studies showed no protective effect (Legendre et al. 1990; Hoover et al. 1991). A second version of this vaccine (Leukocell 2) containing higher concentrations of viral proteins has subsequently been released. This vaccine has been reported to have improved effectiveness (Pollock and Haffer 1991; Tizard and Bass 1991). However, another study failed to find protection (Legendre et al. 1991).
Other commercial FeLV vaccines have also reached the market, including whole inactivated viral preparations (Hines et al. 1991; Hoover et al. 1991; Legendre et al. 1991; Pedersen and Johnson 1991; Tizard and Bass 1991; York and York 1991; Pedersen 1993) and vaccines consisting of recombinant gp70 (SU) protein (Clark et al. 1991; Kensil et al. 1991; Lehmann et al. 1991; Marciani et al. 1991). Although some disagreement exists, these vaccines generally have been effective in preventing challenge infection, particularly when inhibition of persistent infection is used as the criterion of success. In some cases, the efficacy of the vaccine preparations was correlated with the capacity to induce neutralizing antibodies in vaccines, which would support a role for humoral immunity in protection (Hoover et al. 1991). There are studies, however, in which vaccinated cats have resisted challenge infection without developing neutralizing antibodies prior to challenge (Pedersen 1993). This indicates that other forms of immunity (presumably cellular) are important in resisting infection.
It should be noted that many unvaccinated FeLV-infected cats clear initial FeLV infection, as measured by lack of persistent viremia. Thus, FeLV vaccines may act to reduce the initial viral burden during a challenge infection to levels low enough to allow most animals to eliminate the infection. Indeed, in many FeLV vaccine studies, transient expression of the challenge virus is observed (Clark et al. 1991; Hines et al. 1991; Pedersen and Johnson 1991).
A vaccinia recombinant expressing FeLV gp70 (SU) has been tested, but it did not elicit neutralizing antibodies and did not protect against viral challenge (Gilbert et al. 1987). However, a similar canarypox recombinant expressing the entire FeLV gag and env genes protected cats from viral challenge (Tartaglia et al. 1993). In this case, virus-specific neutralizing antibodies were not detected; presumably virus-specific CTLs were responsible for the protection. A live recombinant feline herpesvirus expressing FeLV Env or Gag proteins induced protection from challenge infection, but only if the vaccines were boosted with recombinant Env and Gag proteins (Wardley et al. 1992).
The collective experience with FeLV vaccines is that several different approaches, including inactivated whole virus and Env subunits, have been successful. Moreover, success has been achieved under field conditions in outbred animals. It is noteworthy that in at least some cases, induction of detectable neutralizing antibodies was not a prerequisite for protection.
Bovine Leukemia Virus
Bovine leukemia virus (BLV), which is related to human T-cell leukemia virus (HTLV-1 and HTLV-2), causes lymphosarcoma in cattle with a long latent period. BLV is more pathogenic in sheep, and many experimental studies have been carried out in this species.
Immunological Responses in BLV-infected Animals
BLV-infected animals produce virus-specific antibodies, including those with neutralizing activity (Bruck et al. 1984). The neutralizing determinants have been mapped to the amino terminus of gp51 (SU) (Bruck et al. 1984; Mamoun et al. 1990; Callebaut et al. 1993). Cellular immunity to BLV has also been detected in infected animals, and CTL epitopes have been mapped on the Env SU protein (Gatei et al. 1993a) as have CD4 (T helper) epitopes (Callebaut et al. 1993; Gatei et al. 1993a).
BLV Vaccines
Various vaccine approaches have produced resistance to challenge infection. The first successful vaccine consisted of fixed BLV-infected cells (Onuma et al. 1984). Purified SU could also induce protective immunity (Onuma et al. 1984). Other investigators have achieved protective immunity with inactivated virus (Fukuyama et al. 1993) or with live BLV-infected cells that do not produce infectious virus (Altaner et al. 1991).
Vaccinia virus recombinants expressing BLV gp51 (SU) (Ohishi et al. 1988; Gatei et al. 1993b) or gp51 and gp30 (TM) (Portetelle et al. 1991) protect sheep against BLV infection and disease. Polymerase chain reaction (PCR) has been used to detect BLV proviral DNA in vaccinated and challenged animals. BLV DNA can be detected shortly after challenge, but after long periods of time, viral DNA is undetectable (Gatei et al. 1993b). Challenge virus is cleared in vaccinated animals but not in unvaccinated controls. In one study, vaccinated sheep did not develop virus-neutralizing antibodies, which suggests that cellular immunity may be sufficient for protection (Ohishi et al. 1988). In another study, the degree of protection correlated with development of a CD4+ T-helper-cell response against a gp51 CD4 epitope (Gatei et al. 1993b). On the other hand, passive antibody administration by injection of BLV-neutralizing antiserum has also been reported to provide protection from a challenge infection (Kono et al. 1986).
Human T-cell Leukemia Virus
Some vaccine studies have been carried out on HTLV-1. A panel of monoclonal antibodies developed from HTLV-1-infected individuals has been used to identify a major neutralization domain in env gp46 (SU) protein (Baba et al. 1993). HTLV-1 can productively infect rabbits with low efficiency, and poxvirus recombinants expressing HTLV-1 Env can induce protective immunity to HTLV-1 challenge in rabbits (Shida et al. 1987; Franchini et al. 1995a). HTLV-1 proteins shed from an infected human T-lymphocyte line have been used to immunize pigtail macaques (Dezzutti et al. 1990a,b). This resulted in protection from challenge infection by the related simian T-cell lymphotropic virus (STLV-1). Bacterially expressed HTLV-1 Env protein was able to protect cynomolgus monkeys against challenge infection by HTLV-1 (Nakamura et al. 1987). A candidate peptide vaccine carrying B- and T-cell epitopes from HTLV-1 Env has been described (E. Baba et al. 1995).
CTLs specific for Tax protein are frequently detected in humans infected with HTLV-1 (Jacobson et al. 1990; Parker et al. 1992). In some studies, these CTLs are associated with the disease tropical spastic paraparesis (TSP; see Chapter 10 (Jacobson et al. 1990), but other workers have not seen this correlation (Parker et al. 1992, 1994).
Simian Type-D Retrovirus
Simian type-D retrovirus induces an immunodeficiency syndrome in rhesus macaques that resembles AIDS. Prior to the isolation of SIVs, this virus received attention as a primate model for AIDS. A formalin-inactivated whole-virus vaccine was effective at preventing challenge infection (Marx et al. 1986), and a recombinant vaccinia virus vaccine expressing the viral Env protein was also able to induce protection from low-dose challenge infection (Hu et al. 1989).
Feline Immunodeficiency Virus
Feline immunodeficiency viruses (FIVs) are lentiviruses that infect a significant fraction of domestic cats, as well as some large feral cats (e.g., lions). There has been considerable interest in FIV as a model for HIV, since it induces immunodeficiency in domestic cats. Vaccines consisting of inactivated-virus-infected cells or inactivated purified virus are protective against challenge by homologous or heterologous FIV strains (Yamamoto et al. 1993; Hosie et al. 1995). For these vaccines, protection correlated with the generation of neutralizing antibodies, and sera from vaccinated cats could passively protect naive cats from challenge FIV (Hohdatsu et al. 1993). Although these results indicate that neutralizing antibodies are important in protection against viral challenge, a role for cell-mediated immunity has not been ruled out. Synthetic peptides have been used to raise anti-FIV-specific CTLs in cats, although challenge experiments have not yet been reported (Flynn et al. 1994). In a cautionary note, administration of Env subunit vaccines has been found to enhance FIV infection following challenge (Siebelink et al. 1995).
EIAV, Visna/Maedi Virus, and Caprine Arthritis Encephalitis Virus
The lentiviruses of large ungulates are relatively closely related to HIV. EIAV is particularly noteworthy in its high frequency of antigenic variation within an infected host.
Immunological Responses to Infection
EIAV causes repeated bouts of acute infection in infected horses. Each bout is limited by the appearance of neutralizing antibodies that bring the infection under control. A new bout results from outgrowth of a variant virus whose replication is not blocked by the existing antibodies; this cycle can occur repeatedly (see Chapter 10. As expected, the viral variants have changes in the env gene, and there is no set pattern of variations when the same virus is introduced into different hosts (Montelaro et al. 1990). The variation in the env gene is the result of selection imposed by the neutralizing antibodies produced by the host. This observation illustrates the formidable challenge in developing vaccines against retroviruses with this type of antigenic variation. Infected animals develop antibodies to both conserved and variable regions of the env SU (gp90) protein; the principal neutralizing determinant is in the variable region (Hussain et al. 1988; Ball et al. 1992). Virus-specific cell-mediated immunity has not been extensively studied in EIAV-infected horses, although evidence for some kind of cellular immunity has been obtained using cell proliferation assays (Issel and Coggins 1979).
Like EIAV, visna virus induces neutralizing antibodies in infected sheep (Clements et al. 1988), and visna also shows antigenic variation in animals. In contrast, caprine arthritis encephalitis virus (CAEV) does not induce neutralizing antibodies in infected animals, although nonneutralizing virus-specific antibodies do develop (Clements et al. 1988). As a result, CAEV does not show the high-frequency mutation in the env gene that EIAV and visna do.
Vaccines
Early work indicated that an inactivated EIAV preparation could protect naive horses from the same virus, but not from other isolates (Kono et al. 1970). More recently, an inactivated whole-virus vaccine has been shown to protect against challenge infection by the homologous EIAV strain (Issel et al. 1992). On the other hand, infection by a heterologous challenge virus was not prevented, although viral load and disease were reduced. A subunit vaccine of EIAV Env protein also protected against a challenge with homologous, but not heterologous, EIAV. Moreover, a challenge virus from a heterologous strain enhanced symptoms of the disease.
Simian Immunodeficiency Virus
Because of its close relationship with HIV, SIV is an important model system for HIV vaccine development. Moreover, several SIV strains induce AIDS when infected into primates, particularly in nonnatural host species (e.g., SIVmac in rhesus macaques; see Chapter 11. In contrast, the major nonhuman primate infectable by HIV-1 is the chimpanzee, which limits the number of animals available for experimentation and greatly increases the cost; in addition, HIV-1-infected chimpanzees do not develop immunodeficiency. Given these limitations, considerable work has been done to test the ability of candidate SIV vaccines to prevent infection and/or disease.
Immunological Responses to SIV Infection
Several approaches have been used to map the epitopes recognized by antibodies produced by SIV-infected animals. Antibody responses against viral Env proteins, gp130 (SU) and gp41 (TM), have been the predominant focus of these mapping efforts (see also Chapter 3. Overlapping peptides corresponding to Env proteins have been tested for their reaction with sera from infected (or vaccinated) animals (McBride et al. 1993; Samuelsson et al. 1993). These peptides detect antibodies that react with linear epitopes, but they will not detect antibodies that react with discontinuous epitopes. This approach identified as major linear epitopes several hypervariable regions of gp130 (V1, V2, and V3), the carboxyl terminus of gp130, and conserved regions in gp41. In general, these regions correspond to immunoreactive regions of the HIV-1 Env proteins. As expected, some of the immunoreactive determinants lie within type-specific regions of the Env protein and others are group-specific (M.A. Miller et al. 1992). Group-specific determinants are clearly of more interest in vaccine development.
Considerable effort has been focused on regions of the SIV Env proteins recognized by neutralizing antibodies (neutralization epitopes). Peptides or portions of Env protein were tested for the ability to adsorb neutralizing antibodies from the sera of infected animals. The major neutralization epitope for SIVmac (probably type-specific) is located in the carboxy-terminal third of gp130 but does not correspond to the V3 region of HIV-1 (which is the principal neutralizing determinant for that virus) (Javaherian et al. 1992; Torres et al. 1993). A conformation-dependent group-specific neutralization epitope has also been identified (Haigwood et al. 1992; Choi et al. 1994).
As mentioned above, antibodies that enhance retroviral infection are of concern in the development of vaccines. Antibodies that enhance the infectivity of SIV in vitro have been detected in macaques vaccinated with inactivated whole SIV, but the presence of these enhancing antibodies did not correlate with the ability of the animals to resist infection by a challenge with live SIV (Montefiori et al. 1990). On the other hand, the levels of enhancing antibodies in SIVmac-infected rhesus macaques increase during the course of infection and peak immediately prior to death (Robinson and Mitchell 1990). Although the importance of enhancing antibodies in in vivo infections remains to be established, it may be advisable to eliminate enhancing epitopes from candidate vaccines. The predominant regions that generate enhancing antibodies for SIV are located in the Env (TM) protein, and these sequences are conserved in the TM proteins of HIV-1 and HIV-2 (Robinson et al. 1991). Antibody enhancement for SIV has been attributed to the binding of virus to antibody-coated virus complement receptors on the cell surface.
Virus-specific CTLs have been detected in SIV-infected rhesus macaques (Yamamoto et al. 1990a). These CTLs are predominantly directed against the Env protein, although Gag-specific CTLs have been detected as well (Yamamoto et al. 1990b, 1992). The Gag-specific CTLs were directed against a localized region of gag, and these CTLs were detected only in monkeys bearing a particular MHC class I allele (Yamamoto et al. 1990b; Miller et al. 1991). This is not surprising, since CTLs recognize antigens as processed peptides held by MHC class I molecules.
CD4+ immune responses (predominantly T helper) to SIV Gag and Env proteins have also been detected in macaques immunized with inactivated SIV or preparations of virus-like particles (VLPs) (Mills et al. 1991; Jones et al. 1992). In contrast to the Gag-specific CTL response, the Gag-specific CD4+ response was directed against multiple regions of Gag. CD4+ cells recognized epitopes from the SIV Env protein, although it appeared that animals with different MHC class II genotypes might recognize common epitopes. It might be useful to include such determinants in candidate vaccines.
Vaccines
SIV vaccines have been tested extensively. Most of the work has involved the SIVmac 239 or 251 strains (and their derivatives) since these strains cause AIDS relatively rapidly in rhesus macaques; symptoms typically occur within 1–2 years but with a wide range. The standard criterion has been the development of sterilizing immunity: complete resistance of vaccinated animals to challenge infection. Viral subunit vaccines have not been effective in inducing this type of resistance to a challenge infection. On the other hand, several investigators have shown that vaccination with inactivated SIV could induce protective immunity (Desrosiers et al. 1989; Murphey-Corb et al. 1989; P.R. Johnson et al. 1992). However, there is a problem with these results. SIV is usually grown in human T-cell lines, since few simian T-cell lines exist that are suitable for viral propagation. Vaccinated animals were found to develop strong immunological responses to cellular proteins from the human T-cell lines (Stott 1991; Langlois et al. 1992). When the vaccinated animals were subsequently challenged with SIV grown in monkey cells, most were infected (Le Grand et al. 1992; Cranage et al. 1993; Putkonen et al. 1993), and it is now generally believed that the reports of success with inactivated SIV vaccines did not represent induction of true antiviral immunity. However, some inactivated SIV vaccines may produce partial protection from monkey-cell-grown SIV (Stahl-Hennig et al. 1993).
The only approach that has been reported to produce effective protective immunity is vaccination with an attenuated SIV. Deletion of the nef gene from SIV generates a replication-competent virus that grows poorly in vivo and is nonpathogenic (Kestler et al. 1991). Infection of adult macaques with a nef – mutant of SIV induced protective immunity to high-dose challenge by a wild-type pathogenic SIV (Daniel et al. 1992). This attenuated virus is expected to induce both antiviral humoral and cellular immunity, which might be responsible for its efficacy. The major problem with developing an attenuated vaccine for clinical use is safety. Attenuated viral vaccines have occasionally given rise to pathogenic revertants in vaccines (e.g., in the Sabin poliovirus vaccine). The high mutation rates and recombination frequencies of retroviruses make this a serious consideration; indeed, restoration of partial nef deletions has been observed in SIV-infected macaques (Dittmer et al. 1995; Whatmore et al. 1995). Moreover, it has recently been found that the nef – SIV causes immunodeficiency when the virus is introduced into newborn macaques, which raises concerns about the safety of this particular attenuated virus (Cohen 1994a; T.W. Baba et al. 1995). Two other studies with different attenuated SIV vaccines have also shown protection against challenge infection in vivo (Almond et al. 1995, Clements et al. 1995). In one study, protection from heterologous SIV challenge was correlated with cross-reacting neutralizing antibodies (Clements et al. 1995). In a related approach, infection of cynomolgus monkeys with HIV-2 (which does not cause disease) resulted in long-term protection (>5 years) against immunodeficiency after challenge with pathogenic SIVsm (Putkonen et al. 1995). Although HIV-2 infection did not completely prevent SIVsm infection, the viral loads of the challenge virus were reduced (see also below).
Subunit vaccines for SIV have also been tested. Injecting serum from SIVsm-infected monkeys into naive recipients can produce short-term passive protection from SIVsm infection (Putkonen et al. 1991a). This indicates that neutralizing antibodies (predominantly against Env protein) may be sufficient to protect against infection. Several different approaches using Env protein to induce immunity have been tested. Purified gp130 (SU) protein can induce partial protection (Murphey-Corb et al. 1990, 1991). A vaccinia recombinant expressing gp130 has also been generated, and it confers partial protection (as indicated by reduction in viral load), particularly if vaccinated animals are subsequently inoculated with purified gp130 protein prior to challenge (Giavedoni et al. 1993). Although neutralizing antibodies were induced by the vaccines used in these studies, this protocol does not produce sterilizing immunity. On the other hand, work with a less pathogenic SIVmac strain (MNE) has been somewhat more encouraging. Vaccination of monkeys with recombinant proteins representing different SIV Env domains resulted in partial or complete protection from live SIV challenge, and the animals did not show signs of progression to AIDS (Shafferman et al. 1991). Moreover, these animals initially showed evidence of SIV infection after challenge, but at later times, infectious virus could not be recovered from the vaccinees (Shafferman et al. 1993).
SIV vaccines that induce cellular immunity are also under development. As discussed above, induction of virus-specific CTLs will be challenging, since CTL epitopes are likely to differ depending on the MHC class I genotype of the vaccinee. Since the units of recognition are processed antigenic peptides held by an MHC class I molecule, virus-specific CTLs can be raised against both internal and external virion proteins. As described above, CTL responses to both SIV Gag and Env determinants have been detected in infected or immunized monkeys. In developing an anti-SIV CTL vaccine, it may be preferable to target Gag rather than Env, since there is less sequence variation in Gag than in Env. Infection of monkeys with a vaccinia recombinant expressing SIV Gag has been shown to induce CTLs directed against the same Gag CTL epitope recognized by CTLs in SIV-infected animals (Shen et al. 1991). Subsequent boosting of such animals with a particulate preparation of VLPs containing a fusion between SIV Gag protein and Gag-like protein from the yeast Ty1 retroid element also elicited a strong CTL recall response (Shen et al. 1993).
Monkeys have been injected with a synthetic peptide corresponding to the MHC class-I-specific Gag CTL epitope (M.D. Miller et al. 1992). Incorporation of the peptide into an artificial membrane vesicle (proteoliposome) allowed the peptide to be presented by the MHC class I complex without de novo synthesis in the antigen-presenting cell. Gag-specific CTLs were detected in animals carrying the corresponding MHC class I allele. However, induction of CTLs to this Gag epitope was not sufficient to protect animals from a challenge infection (Yasutomi et al. 1995). It should be noted that in order for this approach to be useful as a vaccine in outbred animals, strong CTL epitopes for many different MHC class I genotypes must be identified and the corresponding protective peptides must be included in the vaccine.
As mentioned above, the standard criterion for success in these experiments has been the development of sterilizing immunity. By this criterion, most of the SIV vaccine studies have been unsuccessful. However, recent results have raised the question of whether sterilizing immunity is the most relevant measure of success. The critical issue is whether vaccination prevents virus-induced pathology. As mentioned in the introduction, most viral vaccines now in use do not completely inhibit challenge viral infection, although they prevent disease. It has recently been reported that some monkeys vaccinated with various SIV vaccines show delayed progression to AIDS upon challenge infection with pathogenic SIV, even though there was infection with the challenge virus (Shafferman et al. 1991, 1993; Hirsch et al. 1994). In addition, as described above, immunization of cynomolgus monkeys with HIV-2 prevented AIDS after infection with SIVsm even though low-level infection with the challenge virus occurred (Putkonen et al. 1995). It will be important to retest some of the existing SIV vaccines for efficacy in preventing or delaying progression to AIDS after challenge infection.
An important issue in the development of SIV (and HIV) vaccines is generation of mucosal humoral immunity. Mucosal immunity (and production of antibodies of the IgA class) is likely to be important in preventing infection following vaginal and rectal exposure. The route of administration of the vaccine has been shown to influence the development of mucosal immunity. Vaginal plus oral or ororectal administration of an SIV vaccine consisting of yeast Ty1 VLPs containing SIV p27 Gag protein leads to IgA and IgG class antibodies in vaginal or rectal secretions (Lehner et al. 1992, 1993, 1994). In contrast, intramuscular injection of the same vaccine leads only to the production of circulating antibodies. It has also been shown that intramuscular injection plus oral administration of an inactivated whole SIV vaccine encapsulated in microspheres can confer protection to low-dose vaginal challenge by SIV. Administration of an attenuated SIV strain on the vaginal mucosa has also been shown to induce SIV-specific CTLs and antibodies (IgG and IgA) (Lohman et al. 1994).
Human Immunodeficiency Virus
Development of an HIV-1 vaccine has been the subject of intense research ever since the original isolation of HIV-1. Although some candidate vaccines are undergoing phase I clinical trials (establishing safety and appropriate doses), development and certification of an effective vaccine are a difficult problem. The problem is complicated by the fact that in contrast to what has been done previously, in which vaccines were used only prophylactically (to prevent an initial infection), attempts are now being made to develop HIV-1 vaccines that can be used therapeutically to treat already infected patients. The intention, with a therapeutic vaccine, is to stimulate the host's immune system so that it can more effectively interfere with viral replication. This necessarily implies that the host's immune response is somehow suboptimal and that increasing or enhancing the response will be clinically useful. This is quite different from the goal of a prophylactic vaccine, where the intent is not to change the ability of the host to respond, but to prepare the immune system to respond immediately to an initial infection. It should be clear that the development of a therapeutic vaccine for HIV-1 will be a difficult task not only for all the reasons it will be difficult to develop a prophylactic vaccine, but also because recent studies show that the host mounts a strong and vigorous response to HIV-1 infection. The initial reduction in viral load, which takes place in the first few weeks following infection, is the result of the host's immune response (for more details, see Chapter 11. Moreover, there is reason to believe that protocols which stimulate T-cell proliferation can enhance the replication of HIV-1. Under these circumstances, it will be necessary to proceed cautiously and to keep the different goals and requirements of immunological strategies intended for the treatment and the prevention of HIV-1 infection clearly in mind.
Several important issues complicate all types of HIV-1 vaccine research. The only animal model in which HIV-1 replicates efficiently is the chimpanzee, but these animals do not develop AIDS. As described above for SIV, it is unclear whether sterilizing immunity (complete protection from challenge infection) is an appropriate criterion for efficacy of a prophylactic HIV vaccine. As discussed below, none of the vaccines currently in clinical trials are likely to induce sterilizing immunity. Since HIV-1 has a long latent period (typically 10 years or more), it will be time consuming to test both the safety of candidate vaccines and their ability to prevent disease. Safety of HIV-1 vaccines is an important issue: Reversion to pathogenicity is a problem for live attenuated viruses, and vaccines that produce enhancing antibodies might actually potentiate infection. Finally, since the standard clinical diagnosis for HIV-1 infection is the presence of antibodies, it will be necessary to develop methods that can distinguish an individual who has been vaccinated from one who is actually infected.
Immunological Responses to Infection
The immune response to HIV-1 infection has been extensively studied in human patients, and antigenic determinants on HIV proteins have been characterized in various animal systems. These data have been reviewed elsewhere (Putney and Bolognesi 1990). The major focus here is on the production of anti-HIV neutralizing antibody and the cellular immune responses in infected individuals, since these are the most relevant to vaccine design.
Humoral immune responses
Neutralizing antibodies against HIV-1 have been detected in infected individuals, particularly during the asymptomatic phase (see Chapter 11. The majority of these antibodies are directed against the envelope proteins gp120 (SU) and gp41 (TM). This is the expected result, since these proteins are on the exterior of the virion where neutralizing antibodies can bind to them. The regions of the Env proteins recognized by antibodies are summarized in Figure 9 (see also Chapter 3. The principal neutralizing determinant (PND) in gp120 has been mapped to the third variable region (V3) of the protein (Palker et al. 1987; Goudsmit et al. 1988; Rusche et al. 1988; Kenealy et al. 1989). This segment is thought to correspond to a loop of amino acids in the protein (see Chapter 3. Unfortunately, neutralizing antibodies against the V3 loop are generally type-specific; this is a region of the Env protein that is highly variable in different isolates. Other weaker neutralizing determinants in gp120 are recognized by antibodies produced in HIV-infected individuals. An important determinant is in the region involved in binding the receptor protein CD4. Antibodies that block CD4 binding are directed against the carboxy-terminal third of gp120 and in general are able to cross-react with different HIV strains (Berkower et al. 1989; Javaherian et al. 1989; Ho et al. 1991; Steimer et al. 1991). The CD4-binding epitope on gp120 is conformation-dependent; i.e., discontinuous segments of the protein are involved in forming the epitope.
Neutralizing antibodies directed against gp41 have also been detected in HIV-infected individuals (Berkower et al. 1989; Muster et al. 1993). These antibodies recognize an amino-terminal segment on the exterior of the virion, and also an internal segment. A portion of the external gp41 domain is a dominant neutralization determinant, and this region is generally conserved between different HIV strains (Muster et al. 1993).
HIV-specific antibodies have been detected that can enhance in vitro infectivity. These antibodies may promote infection through the binding of virus-antibody complexes to Fc receptors (Homsy et al. 1989) or complement receptors (Robinson et al. 1991) on target cells (e.g., macrophages). A monoclonal antibody to the V3 loop that shows Fc receptor-mediated enhancement of infectivity for an HIV-1 isolate for which it has weak affinity can neutralize an HIV-1 strain to which the antibody binds tightly (Kliks et al. 1993). This raises a concern for vaccine development: Induction of low-affinity antibodies to HIV-1 might enhance rather than inhibit infection, a problem compounded by the variability of the different HIV-1 strains. Unlike Fc receptor-mediated enhancing antibodies, complement receptor-mediated enhancing antibodies are directed against two domains of gp41 (Robinson et al. 1991). The importance of antibody enhancement in HIV infection in vivo is still unclear. In one clinical trial of a recombinant gp160 vaccine, neutralizing antibodies were detected in vaccines, but no enhancing antibodies were detected (Haubrich et al. 1992). On the other hand, experiments with SIV vaccines have shown that induction of antibodies to a region of SIV TM protein known to contain enhancing epitopes reduces the ability of an SIV Env vaccine to protect monkeys for challenge infection (Mitchell et al. 1995).
It is possible that neutralizing antibodies can be raised against HIV Gag, particularly matrix (MA) protein, which is associated with the viral envelope (although it is generally viewed as lying under the envelope lipid bilayer). Antibodies raised against a peptide of human α-thymosin (HGP-30) that has residues in common with HIV-1 p17 (MA) protein can neutralize HIV-1 in vitro (Naylor et al. 1987), and on the basis of this result, HIV-1 p17 protein has been considered to be a target for generation of vaccine development.
Cellular immune responses
CTL responses and T-helper responses may both be important in controlling HIV-1 infection. Several different assays have been used to detect cellular immune responses involving either virus-specific CTLs or T-helper cells or general virus-specific T-cell responses (where CTL and T-helper activities are not distinguished). The presence of virus-specific CTLs is indicated by the killing of MHC-compatible target cells expressing different HIV-1 proteins when lymphocytes from an infected (or vaccinated) individual are mixed with them in vitro. T-helper cell responses can be measured in vitro. Cells taken from the individual being tested are stimulated with viral antigens. If the cells recognize the antigens, they will release IL-2. General cell-mediated immunity is detected by growth stimulation (monitored by measuring the incorporation of labeled thymidine) of cells from an infected individual after the cells are exposed to viral antigens in vitro.
Virus-specific CTLs have been detected in many HIV-1-infected individuals (Walker et al. 1987, 1988; Koenig et al. 1988). Indeed, recent evidence suggests that virus-specific CTLs may be important in controlling the initial acute phase of infection, since viral load is decreased when CTLs first appear (Koup et al. 1994). In contrast, neutralizing antibodies are not detected until considerably later (Safrit et al. 1994). In infected individuals, the most frequent CTL responses are against the Env protein, followed by RT and Gag (Walker et al. 1987, 1988; McChesney et al. 1990). CTLs against Nef have also been detected (Culmann et al. 1989).
Identification of CTL epitopes for different viral proteins is complicated by the fact that the epitopes differ in individuals with different MHC genotypes (see above). The epitope framework sequences are known for many class I alleles (Harding and Unanue 1990), and HIV epitopes for these alleles can be predicted. Human CTLs that recognize Env segments near the V3 loop and the CD4-binding regions have been detected (R.P. Johnson et al. 1992). In Gag, CTL epitopes have been detected in conserved regions of p24 (capsid, CA) protein (Buseyne et al. 1993) and p17 (MA) protein (Achour et al. 1990). In the case of CTLs that recognize CA, the same regions can be recognized in individuals with different MHC class I genotypes (Buseyne et al. 1993), which is encouraging for efforts to design vaccines that will elicit CTL responses in the human population. CTL responses to Pol protein have been mapped to several domains of the RT protein (Walker et al. 1989; Hosmalin et al. 1990). Nef-specific CTL responses may be targeted against the same Nef domain in different infected individuals (Culmann et al. 1989).
General cellular immune responses to the Env precursor protein gp160 have been mapped in HIV-infected individuals by testing for proliferation after stimulation with synthetic gp160 peptides (Berzofsky et al. 1991). This approach indicated that a majority of HIV-infected individuals exhibited T-cell responses to one or more of a small number of gp160 peptides, and 77% of seropositives responded to a mixture of these peptides. These peptides should represent domains that would elicit T-cell responses in individuals with a spectrum of MHC genotypes. A specific T-helper epitope recognized by infected individuals of several different MHC class II genotypes has been mapped in RT (De Groot et al. 1991).
HIV-1 Vaccine Approaches
Many different kinds of HIV vaccines are currently under development. In terms of potential application in humans, those that have received the most attention include subunit vaccines (predominantly for Env proteins), inactivated virus, and vaccinia-based live vaccines. The current experience with these vaccines is summarized in Table 2. Assessing the efficacy of these vaccines is a major challenge. Current criteria for effectiveness include prevention of challenge infection in chimpanzees and the production in humans of neutralizing antibodies or other immune responses against diverse HIV isolates. However, the appropriateness of either of these criteria is open to question. Chimpanzees are more resistant to HIV-1 infection than humans, and in general, unvaccinated chimpanzees will eventually clear an infection. As discussed above, it is unclear if neutralizing antibodies will be able to confer protective immunity in humans to HIV-1 strains under field conditions.
Table 2
Some Current HIV-1 Vaccines Under Development.
Recombinant protein subunit vaccines
These vaccines consist of recombinant viral proteins, predominantly the Env protein, expressed using baculovirus vectors or in mammalian cells. Vaccines consisting of the entire gp160 Env precursor protein (which contains both the SU and TM) or of just the gp120 (SU) domain have been generated. Recombinant gp120 and gp160 corresponding to the original IIIB (LAV/BRU) isolate, as well as the MN isolate (more typical of North American field isolates) are under development. Phase I pilot studies of a gp160 (IIIB) vaccine in HIV seronegative human volunteers indicated that it induces type-specific but not group-specific neutralizing antibodies in some vaccinees (Dolin et al. 1991; Gorse et al. 1992; Kovacs et al. 1993). Group-specific cellular immunity and CD4+ CTLs have been detected (Orentas et al. 1990; Tacket et al. 1990; Clerici et al. 1991; Kovacs et al. 1993; Stanhope et al. 1993). However, gp160 (IIIB) has been unable to prevent challenge infection with homologous virus in chimpanzees (Berman et al. 1990).
Similarly, gp120 (IIIB) can induce neutralizing antibodies for homologous and heterologous HIV-1 strains in humans (Schwartz et al. 1993). In contrast to gp160 (IIIB), gp120 (IIIB) is able to induce protective immunity to low doses of homologous challenge virus in chimpanzees (Berman et al. 1990) but not to higher doses (Berman et al. 1988). Animal model experiments also suggest that gp120 (MN) may be more efficient at inducing antibodies that neutralize more diverse HIV-1 strains. On the other hand, immunization with a combination of gp160 (MN) and synthetic peptides from the V3 loop (MN) resulted in protection of chimpanzees from challenge by a heterologous HIV-1 of the same subtype (SF2) (Girard et al. 1995). The question of whether neutralizing antibody can prevent HIV-1 infection has been at least partially answered by the demonstration that simultaneous inoculation of a neutralizing monoclonal antibody specific for the V3 loop along with homologous challenge virus can inhibit infection (Emini et al. 1992). Neutralizing serum from cynomolgus monkeys vaccinated with inactivated HIV-2 prevented challenge infection by HIV-2 or SIVsm in cynomolgus monkeys when the serum was administered shortly before infection (Putkonen et al. 1991a).
So far, several different gp120 and gp160 vaccines have been tested in phase I clinical trials. The goal of the phase I trials (conducted in uninfected individuals) was to test the safety of the vaccines and to establish the levels of vaccine necessary for production of an antibody response. In total, approximately 1000 individuals have been tested in phase I trials of gp120 and gp160 vaccines in the United States. However, plans to expand the phase I human trials to phase II (efficacy) trials had to be postponed when it was learned that although sera from vaccinees could neutralize diverse laboratory isolates of HIV-1 in vitro, they uniformly failed to neutralize HIV-1 field isolates grown in primary human peripheral blood mononuclear cells (Cohen 1993; Mascola et al. 1996). This makes it quite unlikely that the current gp160 and gp120 vaccines will be effective in preventing HIV-1 infection in field trials. In light of this result and of the fact that several individuals who have received these vaccines have subsequently been infected with HIV-1 (Cohen 1994b; Kahn et al. 1995; Mascola and McNeil 1995), large-scale trials of these vaccines in the United States have been postponed indefinitely. However, plans are being made to test gp160 or gp120 vaccines in other parts of the world where the rate of HIV-1 infection is extremely high (Moore and Anderson 1994).
The use of gp160 to stimulate antiviral immunity in HIV-infected individuals has also been considered. In one small study, administration of gp160 to asymptomatic HIV-infected individuals resulted in increases in specific CD4+ and CD8+ CTLs (Kundu et al. 1992). In a larger study, the CD4 cell counts were studied in asymptomatic infected individuals inoculated with gp160 (Redfield et al. 1991). Individuals whose antiviral immune responses (humoral or cellular) increased in response to gp160 administration showed no changes in CD4 count during a 10-month period, whereas those who did not respond showed a small but measurable decrease in CD4 count. These results might be interpreted as suggesting that the gp160 vaccine benefited the HIV-1-infected subjects, but it was not clear whether the inoculation with gp160 caused the stabilization of CD4 counts in the responders or if the immune systems were more intact in the responders. Large, long-term trials (3-year and 5-year) of gp160 administration to asymptomatic HIV-1-infected individuals have recently been completed, and no clinical benefit or delay in disease progression was observed (Kaiser 1996).
A vaccine targeted at p17 (MA) Gag protein has also been tested in humans (Kahn et al. 1992). This vaccine is based on the observation that antisera against a fragment of p17 (HGP-30) can neutralize HIV-1. Preliminary studies showed that administration of HGP-30 to humans resulted in low-level antibody responses and potentially in the production of p17-specific CTLs.
Inactivated virus
Inactivated whole virus has also been considered as an HIV vaccine. This is a classical approach that has been successful for several other viruses. Protection against homologous HIV-1 challenge infection in chimpanzees has been achieved by immunization with inactivated HIV-1, followed by boosters of peptides corresponding to the V3 loop region of gp120 (Girard et al. 1991). Moreover, animals immunized in this manner are resistant to challenge by cell-associated virus (Fultz et al. 1992), although interpretation of the results must resolve the potential contribution of human class I molecules in viral preparations (see above). Cynomolgus monkeys immunized with inactivated HIV-2 have also been protected from homologous viral challenge (Putkonen et al. 1991b).
Inactivated HIV-1 preparations (largely lacking Env protein) are also being tested as immunotherapeutics in infected individuals. Some preliminary success in inducing antiviral immunity, reducing viral load, and preventing further decline in CD4 cell counts has been reported, although these results have yet to be published. A potential problem with any immunization in HIV-1-infected individuals is that immunization may increase HIV-1 replication by stimulating the immune system. Influenza virus vaccinations have been found to increase the HIV-1 RNA levels in infected individuals (O'Brien et al. 1995).
Vaccinia-based vaccines
Recombinant vaccinia viruses expressing HIV-1 Env (gp160) have been generated. As live vaccines, these viruses have the advantage of inducing both CTLs (CD8+ and CD4+) and neutralizing antibodies. However, initial studies in humans with recombinant vaccinia viruses that produce gp160 resulted in relatively weak antibody and cellular immune responses (Graham et al. 1992). This problem was compounded by the fact that the immune responses to gp160 were even weaker in individuals who previously had received wild-type vaccinia as a vaccine against smallpox—a substantial portion of the adult population (Cooney et al. 1991). Moreover, the vaccinia gp160 vaccine does not confer protective immunity to HIV-1 challenge in chimpanzees (Hu et al. 1987).
Recently, more encouraging results have been obtained by boosting human subjects who had received the vaccinia gp160 vaccine with recombinant Env protein (gp160 or gp120). Such procedures led to the production of CD4+ and CD8+ CTLs and high titers of neutralizing antibodies (Berzofsky et al. 1988; Hammond et al. 1992; Cooney et al. 1993; Graham et al. 1993). In addition, introduction of peripheral blood mononuclear cells from these individuals into SCID mice leads to the propagation of human blood cells that resist challenge infection (Mosier et al. 1993). Immunization with vaccinia virus recombinants expressing HIV-2 sequences followed by boosting with HIV-2 gp160 resulted in efficient protection from challenge exposure to HIV-2 (Franchini et al. 1995b). In addition, administration to humans of a canarypox vector expressing HIV-1 Env resulted in production of HIV-1-specific CTLs (Egan et al. 1995). On the other hand, a canarypox vector expressing HIV-1 Env followed by boosting with HIV-1 gp160 did not lead to protection from heterologous HIV-1 challenge infection in chimpanzees (Girard et al. 1995). Thus, vaccinia (or similar canarypox)-based vaccines still show some promise, particularly when used with viral protein boosters.
Passive immunity
Administration of neutralizing antibodies (passive immunity) might be useful in specific situations, e.g., if they are administered immediately after known exposure to HIV infection. It has been shown that anti-Env monoclonal antibodies can prevent challenge infection by HIV-1 in chimpanzees (Emini et al. 1992). Monoclonal antibodies have been isolated that can neutralize diverse HIV-1 isolates, including laboratory strains and primary isolates (Burton et al. 1994; Conley et al. 1994a,b), and they might be used to prevent HIV-1 infection. Recently, a clinical trial of passive immunotherapy involving administration of plasma from asymptomatic HIV-1-positive individuals to symptomatic infected individuals showed some efficacy in delaying the onset of AIDS symptoms (Vittecoq et al. 1995), and a phase II efficacy trial has been approved.
Novel and experimental vaccine strategies
Other novel vaccine strategies now under development are described in Table 3. These may be important, given the current discouraging situation with the recombinant subunit vaccines.
Table 3
Other HIV-1 Vaccine Approaches.
Several of the approaches are based on immunization with particulate preparations of HIV-1 proteins rather than monomeric proteins. In animal experiments, particulate immunogens are usually more effective in inducing neutralizing antibodies. One class of particles are virus-like HIV-1 particles consisting of Gag protein fused to Env sequences. Expression of high levels of such chimeras (e.g., from cells expressing the chimeric proteins from vaccinia-based vectors) can lead to assembly of VLPs that are released into the culture medium. Such VLPs produce higher titers of neutralizing antibodies and T-helper responses in rabbits than do monomeric proteins (Haffar et al. 1991; Vzorov et al. 1991; Luo et al. 1992). Similarly, fusion proteins containing hepatitis B surface antigen and HIV-1 Env domains assemble into particulate structures that can induce neutralizing antibodies and T-cell responses in rhesus monkeys (Schlienger et al. 1992). VLPs based on chimeras between yeast Ty1 Gag domains and HIV-1 Env (V3) domains have also been shown to induce neutralizing antibodies and T-cell proliferative responses more efficiently in mice than immunizations with monomeric proteins (Griffiths et al. 1991; Harris et al. 1992).
Another approach involves vaccines that replicate within the vaccinee, as do the vaccinia-based vaccines described above. These vaccines should induce both neutralizing antibodies and virus-specific CTLs since the viral proteins are synthesized de novo in cells of the vaccinee. Several different types of viral vectors have been engineered to express HIV-1 proteins. These include vectors based on adenovirus (Natuk et al. 1993), poliovirus (Evans et al. 1989; Ansardi et al. 1991; Porter et al. 1993), rhinovirus (Resnick et al. 1994), and influenza virus (Kalyan et al. 1994; Muster et al. 1994). These vectors have been shown to induce high titers of neutralizing antibodies and/or cellular immune responses in animals. Moreover, intranasal administration in mice of an influenza virus expressing an HIV-1 gp41 conserved epitope resulted in production of HIV-1-specific secretory antibodies (IgAs) in several mucosal tissues (Muster et al. 1995). An important issue for any live vaccine is biosafety. In addition, prior infection or immunization with the virus bearing the HIV-1 proteins (e.g., poliovirus and influenza virus) may reduce the efficiency of infection of these vaccines in some individuals.
Included in the category of replicating-virus-based vaccines are attenuated strains of HIV-1. As described above, a nonpathogenic nef – mutant of SIV was able to prevent SIV challenge infection. Given the relatively dim prospects for the subunit vaccines, development of an attenuated HIV-1 vaccine is receiving more attention, although the safety issue will need to be resolved. In this regard, it has been reported that an HIV-1-infected long-term nonprogresser had been infected with a nef – variant (Kirchhoff et al. 1995), which would support the idea that these strains have reduced pathogenicity. A group of HIV-1-infected individuals in Australia has been identified that were all infected by blood from the same person. None of the people in this group have progressed to AIDS. Recent studies showed that all of these individuals were infected with nef – HIV-1 (Deacon et al. 1995). On the other hand, systematic studies of AIDS patients have not been carried out to determine if a class exists consisting of symptomatic individuals that were infected by the nef – virus. The safety of a nef – HIV-1 vaccine would depend on the demonstrated absence of this latter class.
Another live vaccine is based on the bacterium Bacillus Calmette-Guerin (BCG). BCG engineered to express HIV-1 proteins induces humoral and cellular immunity in experimental animals (Aldovini and Young 1991). BCG is currently employed in some countries as a vaccine against tuberculosis, so it has an established record of safety. A BCG-based vaccine expressing the V3 region of HIV-1 gp120 induced HIV-1-specific CTLs and neutralizing antibodies in mice and guinea pigs (Honda et al. 1995). Moreover, sera from the immunized guinea pigs could prevent HIV-1 infection of human cells in SCID mice (SCID/hu and SCID/huPBL). Although recombinant BCG could potentially be used as a prophylactic vaccine, it will probably not be used as an immunotherapeutic for HIV-infected individuals since systemic BCG infections have been detected in AIDS patients.
Another potential bacterium-based vaccine is one based on Salmonella typhimurium. Salmonella induces potent mucosal and systemic immunity when administered orally. An S. typhimurium-based vaccine expressing HIV-1 gp120 induced specific T-cell responses but not antibody responses in mice (Berggren et al. 1995). On the other hand, a Salmonella vector expressing HIV-2 proteins combined with an HIV-2 gp160 boost did not induce protective immunity in rhesus macaques against an HIV-2 challenge (Franchini et al. 1995b).
A third bacterium-based vaccine now being developed is based on Listeria monocytogenes. L. monocytogenes induces strong cellular immune responses, and an L. monocytogenes expressing HIV-1 Gag protein induces a strong HIV-1 Gag-specific CTL response in mice (Frankel et al. 1995).
Another approach is to use peptides corresponding to antibody or T-cell epitopes as vaccines. This approach has the advantage that the peptides can be chemically synthesized on a large scale, and a cocktail of different peptides corresponding to different B- and T-cell epitopes could be injected simultaneously to induce immune responses to several different viral proteins (Sastry et al. 1992). Peptides might also be used to stimulate cellular or humoral immunity in previously vaccinated or infected individuals (Girard et al. 1991; Hart et al. 1991; Martinon et al. 1992). If peptides are used to induce T-cell responses, the MHC genotype will affect the relative immunogenicity of different HIV-1 peptides as has already been discussed. Another potential difficulty with the use of peptides (and with using purified gp120 or gp160) is that, in general, peptides must be synthesized within target cells if they are to associate with MHC class I proteins and bind specific CTLs. Peptide vaccines would not normally be expected to induce virus-specific (CD8+) CTLs. However, encapsulation of peptides into liposomes or lipid micelles has resulted in association of the peptides with MHC class I proteins and induction of neutralizing antibodies, CTLs, and T-helper lymphocytes in rodent models (Defoort et al. 1992). In animal models, different peptide formulations (with and without adjuvants) have been shown to induce neutralizing antibodies that are cross-reactive to several laboratory strains of HIV-1 (Bukawa et al. 1995; Hamajima et al. 1995); in one case, the formulation resulted in induction of mucosal antibodies (Bukawa et al. 1995). In humans, a branched peptide for the V3 domain of HIV-1 Env (MN) was shown to induce type-specific neutralizing antibody as well as cellular immune responses (Gorse et al. 1996).
A potential solution to the safety issue posed by live vaccines is to use defective retroviral vectors (see also Chapter 9. Like live vaccines, these vectors can induce the expression of HIV-1 proteins in the recipient's cells so that they have a higher probability of inducing virus-specific cellular immunity. MLV-based retroviral vectors that express the HIV-1 Env or Gag induce virus-specific CTLs and neutralizing antibodies in mice (Warner et al. 1991; Chada et al. 1993). Partial induction of protective immunity for SIV infection has been achieved by use of an analogous MLV-based SIV vector. A phase I trial in which such vectors were used as an immunotherapeutic in HIV-1-infected individuals has been completed, and a phase II trial is in progress. The goal is to stimulate virus-specific CTL responses leading to elimination or control of the HIV-infected cells. The initial approach was to cultivate cells from each subject and infect them with a retroviral vector ex vivo. Fixed or killed cells (which can still present HIV antigens) are then reintroduced into the same HIV-infected subject, with the hope that they would stimulate virus-specific CTLs and antibody production. In clinical trials, the more direct approach of injecting retroviral vectors into HIV-1-infected individuals is being used.
A related approach is the direct injection of naked DNA containing sequences designed to express viral proteins. This procedure can cause the expression of proteins encoded by the injected DNA in cells of the vaccinee (see above). Like live vaccines, injection of naked DNA should theoretically induce both virus-specific CTLs and antibodies. Injecting the DNA of an influenza expression vector (described above) will protect mice from lethal challenge by influenza virus. Experiments in mice indicate that injecting naked DNA can induce neutralizing antibodies, T-cell proliferative responses, and CTLs specific for HIV-1 (Wang et al. 1993a,b; Lu et al. 1995; Okuda et al. 1995). More recently, injections of naked DNA have been shown to induce HIV-1-specific neutralizing antibodies and CTLs in macaques (Okuda et al. 1995; Wang et al. 1995; Yasutomi et al. 1996).
In summary, these novel vaccine strategies have potential advantages, although considerable development will be required before it is clear if any of these approaches are superior to more conventional approaches. Ultimately, an effective approach may require the combination of more than one of these vaccine strategies.
Publication Details
Copyright
Publisher
Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY)
NLM Citation
Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997. Immunological Approaches to Controlling Retroviral Infection.








