Introduction
In contrast to immunologic approaches designed to prevent retroviral diseases, pharmacologic approaches designed to treat retroviral infections have focused primarily on the lentiviruses, particularly HIV-1. This is, of course, due to a pressing need for an effective treatment for individuals already infected with this virus. During the past decade, extensive investigations of HIV-1 replication have identified molecular targets suitable for intervention, and a large number of inhibitors have been identified, tested, and developed. In contrast, very little work has been reported with other retroviruses, except as in vivo models for HIV-1 infection. Accordingly, this section concentrates exclusively on HIV-1 and, of necessity, provides only an overview.
Despite much effort, our understanding of the in vivo pathogenesis of HIV-1 is incomplete (see Chapter 11. It was initially assumed that inhibiting viral replication in vivo would result in a decrease in the patient viral burden. Clinical studies in patients with some of the inhibitors described in this section demonstrated that this assumption is correct. HIV-1 infection in vivo is characterized by a high degree of continuous viral replication and by rapid viral turnover (Ho et al. 1995; Wei et al. 1995; Perelson et al. 1996). Therefore, even a relatively modest inhibition of viral replication results in a reduction in the viral burden. In turn, this decreased burden is typically associated with increases in levels of the primary target for HIV-1 infection, the CD4+ T cell. It has yet to be firmly established whether decreases in viral burden and increases in CD4 cell counts will yield long-term clinical benefits. However, a strong correlation appears to exist between viral burden and clinical progression in HIV-1-infected individuals (Mellors 1996). Patients with a lower burden generally exhibit a longer time to progression to AIDS than do patients with a higher viral load.
Lentiviruses provide a number of possible targets for pharmacological intervention (Table 4). Important targets include virion-cell receptor interaction, reverse transcription, integration, proteolytic maturation, and virus-specific transcriptional regulation. Given the incomplete understanding of HIV-1 pathogenesis, it has been difficult to predict, from relatively limited preclinical data, the relative effectiveness of inhibitors directed at these various targets. In addition, a compound that inhibits viral molecular activity in vitro does necessarily lend directly to the development of a practical therapeutic agent. Considerations of toxicity, in vivo pharmacology, and delivery are important.
Table 4
Possible Lentiviral Targets for Pharmacologic Intervention.
An additional and important consideration for the development of agents that can produce long-term clinical benefit is the selection of HIV-1 variants which have a reduced susceptibility to specific inhibitors. The development of resistant viral strains is a problem for all compounds that have exhibited some in vivo antiviral activity and that have been tested in patients.
This has meant, up until recently, that it has not been possible to use drug therapy to reduce the viral load in vivo significantly, and it has not been possible to make definitive conclusions about the long-term clinical benefits of inhibiting HIV-1 replication. Quite recently, certain combinations of drugs have been used that substantially reduce the viral load for more than 1 year in the majority of patients. These results are promising, and we can expect to learn more about the long-term benefits of these therapies from clinical trials now in progress. It is important to emphasize that an understanding has been essential both of the mechanism of action of the more promising inhibitors and of the mechanism of resistance in the development of more effective inhibitors and clinical protocols.
A majority of the anti-HIV-1 drugs in use and in development are inhibitors of RT or protease. As a consequence, this chapter places particular attention on compounds that inhibit these two viral enzymes.
Virion-Cell Receptor Interaction
The primary cell receptor for HIV-1 infection is the CD4 cell surface glycoprotein (Dalgleish et al. 1984; Klatzmann et al. 1984; McDougal et al. 1986). Certain CD4− cells are also apparently infected by the virus, but the overall contribution of these cells to in vivo infection seems to be minimal. Hence, prevention of acute HIV-1 infection by blocking the interaction of CD4 with the viral gp120 envelope glycoprotein has reasonable therapeutic potential.
A soluble recombinant version of the 55-kD CD4 molecule, termed sCD4, was one of the first HIV-1 inhibitors to be extensively studied and tested. The molecule competes with the cell-surface-bound form of the receptor for binding to gp120. sCD4 exhibits some virus-neutralizing activity in cell culture in tests using T-cell-line-adapted variants of HIV-1 (Deen et al. 1988; Fisher et al. 1988; Hussey et al. 1988; Traunecker et al. 1988; Clapham et al. 1989). However, the administration of sCD4 to HIV-1-infected individuals had little or no effect on viral burden (Kahn et al. 1990; Schooley et al. 1990). Subsequent studies in cell culture demonstrated that sCD4 did not have the same neutralizing activity for all of the biological variants of HIV-1 and that it is particularly poor for primary clinical isolates of the virus (Daar et al. 1990). The reason for this difference in activity remains unknown, but it might involve the different coreceptors used by different strains of virus (see Chapter 4). It might also be related to a similar phenomenon with virus-neutralizing antibodies (Moore et al. 1995). These antibodies exhibit potent activity against variants that have been adapted to grow in T-cell lines in culture, but they are notably less effective (or ineffective) against primary patient isolates. It seems that the process of adaptation of the virus to persistent growth in T-cell lines selects for HIV-1 variants that are particularly susceptible to neutralization by antibody (Wrin et al. 1995) and possibly by sCD4 as well.
A second, related approach involved the development of a hybrid molecule composed of the portion of CD4 that binds gp120 and the heavy-chain Fc region of an IgG molecule (Capon et al. 1989). It was hoped that such a hybrid molecule would have the functional characteristics of an antibody and specificity for the CD4-binding site on the viral envelope. The so-called CD4 immunoadhesin molecule was found to have improved pharmacokinetic properties in vivo compared to sCD4. However, clinical studies again failed to demonstrate antiviral activity in patients (Collier et al. 1995; Meng et al. 1995). sCD4 was also fused with the Pseudomonas exotoxin (sCD4-PE40). This toxin fusion was able to specifically kill HIV-infected cells in culture (Chaudhary et al. 1988); however, clinical trials did not show an antiviral effect (Davey et al. 1994).
These negative results do not eliminate the therapeutic potential for a suitably potent, small molecule inhibitor of gp120-CD4 interaction. To this end, the specific regions of CD4 and gp120 involved in their binding are being mapped (see Chapter 3 in the hope that inhibitors can be either designed or discovered by appropriate screening. The structure of CD4 is known (see Chapter 3. There is also a considerable effort to determine the three-dimensional structure of gp120.
Meanwhile, a number of defined molecules that show anti-HIV-1 activity in vitro have been reported that appear to prevent virus binding to CD4. These include aurintricarboxylic acid (Schols et al. 1989), CPF-DD (Finberg et al. 1990), and stilbene disulfonic acids (Cardin et al. 1991). Dextran sulfate and certain other polyanions bind to viral gp120 and interfere with some as yet undefined early event(s) in HIV-1 infection (Schols et al. 1990). Inhibitors of “trimming” glucosidase, such as N-butyl deoxynojirimycin, prevent the appropriate glycosylation of newly synthesized gp120; virions produced in the presence of such inhibitors have a reduced capacity to bind CD4 and are, as a consequence, less infectious (Gruters et al. 1987; Dedera et al. 1990). The therapeutic potential of these compounds is not significant because of unfavorable pharmacokinetics, toxicity limitations, and/or poor potency.
Coreceptors that are apparently required for virus to penetrate cells successfully following initial gp120 interaction with CD4 have recently been identified. These coreceptors are members of a family of related molecules whose normal role is to interact with chemokines, chemotactic cytokines that attract various leukocytes to sites of inflammation. Following the interaction of g]p120 and CD4, a second interaction between the viral envelope glycoprotein and the appropriate chemokine receptor is necessary for gp120 to initiate fusion with the cell membrane. Although the mechanism of this process is not understood, the absolute requirement for interaction with the second receptor makes the receptor a potentially attractive target for the development of a therapeutic intervention. A number of chemokine coreceptors have been identified including CXCR4 (“fusin”), CCR2, CCR3, and CCR5 (Berson et al. 1996; Choe et al. 1996; Deng et al. 1996; Doranz et al. 1996; Dragic et al. 1996; Feng et al. 1996). The relative importance of each of these receptors for the infectivity of HIV-1 patient isolates has not yet been established. CCR5 appears to be particularly important for primary monocytotropic isolates, whereas CXCR4 is an important coreceptor for HIV-1 variants that can infect T-lymphoid cell lines in culture. There are also HIV-1 variants that can use multiple coreceptors. The complexity and diversity of chemokine receptors that interact with HIV-1 may limit the attractiveness of this target for therapeutic development. In any case, considerable further study is required to determine whether any of the chemokine coreceptors will be useful targets.
Reverse Transcription
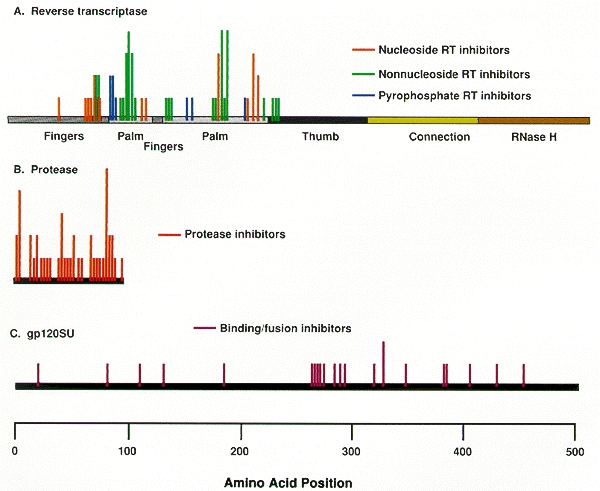
The HIV-1 RT is the most extensively studied viral target for pharmacologic intervention. The enzyme is essential for viral replication since, as with other retroviruses, the expression of HIV-1 proteins and progeny virions requires the conversion of the single-stranded RNA genome in the infecting virion into double-stranded DNA and the subsequent integration of this DNA into the host-cell genome (see Chapters 4 and Chapter 5). Biochemically, RT is an attractive target in that it must complete approximately 20,000 reaction steps to copy the entire 10,000-base RNA genome. Inhibition of any one of these reaction steps results in an aborted infection. Two major classes of anti-HIV-1 inhibitors have been developed and tested in the clinic.
Nucleoside Analog Inhibitors
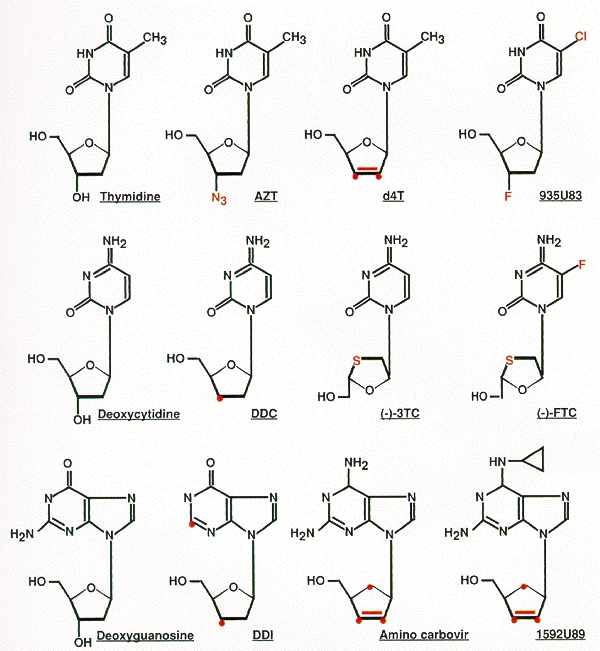
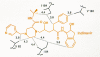
The first class of RT inhibitors is composed of various nucleoside analogs (Fig. 10). Each of these compounds is metabolically activated within cells by phosphorylation. The resulting 5′triphosphates can inhibit the RT enzyme by substrate competition, but the potency of the inhibitors appears to derive from their ability to act as terminators of polymerization chain termination (see Chapter 4 and Mitsuya et al. 1990). RT inhibitors currently licensed for treatment of HIV-1 infection in humans include the nucleoside analogs 3′-azido-3′-deoxythymidine (AZT), 2′,3′-dideoxyinosine (ddI), 2′,3′-dideoxycytidine (ddC), and 2′,3′-didehydro-3′-deoxythymidine (d4T) (Mitsuya et al. 1985; Mitsuya and Broder 1986; Baba et al. 1987; Lin et al. 1987; Browne et al. 1993). Many of these compounds had been originally tested and/or developed as anticancer agents because they can inhibit the DNA polymerases in mammalian cells. Blind screening for RT inhibitory and anti-HIV-1 effects of these “off-the-shelf” compounds identified AZT, ddI, and ddC as potentially useful therapeutic agents.
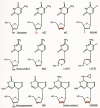
Figure 10
Nucleoside RT inhibitors. Each inhibitor is an analog of the nucleoside shown on the left. Differences from the natural nucleosides are shown in color. Note that 1592U89, shown in the lower right, is a prodrug of amino Carbovir, shown to its left.
When these analogs are used in monotherapy, they have varying degrees of clinical benefit, manifested by some delay in progression of clinical disease and/or by improvement in markers of immune status (Fischl et al. 1987; Yarchoan et al. 1988; Merigan et al. 1989; Cooley et al. 1990; Lambert et al. 1990; Murray et al. 1995; Petersen et al. 1995). Unfortunately, the therapeutic potential of these agents is substantially limited by toxicity (which probably reflects the fact that they do inhibit mammalian DNA polymerases) and by the development of viral resistance (see below).
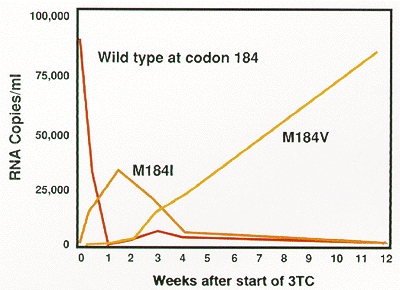
Nevertheless, recent studies, particularly of combination therapies, have shown greater clinical promise for this class of inhibitors. A novel analog, (–)-2′-deoxy-3′-thiacytidine (3TC) (Coates et al. 1992), was developed for use in combination with AZT. 3TC seems to cause less in vivo toxicity than the other nucleoside analogs and, when used with AZT, appears to mediate significant declines in the circulating virus levels and increases in CD4 cell counts (Katlama et al. 1994; Staszewski et al. 1994; Eron et al. 1995). These positive effects are sustained for at least 1 year and may be partially due to a difficulty in selecting for virus that is resistant to both of these nucleoside analogs (see below). The therapeutic regimen of 3TC with AZT was recently granted licensure.
A particularly promising use of the nucleoside analog AZT is in the reduction of maternal-infant transmission. The inhibitor is given to pregnant women in the later stages of pregnancy and to the newborn for 6 weeks after birth. In a comparative trial, AZT treatment reduced the risk of viral transmission almost 70% compared to placebo (Connor et al. 1994). This is the most successful clinical intervention to date for antiretroviral chemotherapy. It also holds real promise for the future; more potent agents (and combinations of agents) that are now becoming available may be even more effective in preventing maternal-infant transmission.
Preclinical and clinical evaluations of additional RT inhibitors that act as chain terminators are in progress. Some of the more advanced studies involve (–)-2′-deoxy-5-fluoro-3′-thiacytidine (FTC) (see Schinazi et al. 1992), 5-chloro-2′,3′-dideoxy-3′-fluorouridine (935 U83) (Daluge et al. 1994), the nucleoside analog 1592U 89 (Saag et al. 1996), and the nucleotide 9-(2-phosphonylmethoxy ethyl) adenine (PMEA) (Pauwels et al. 1988).
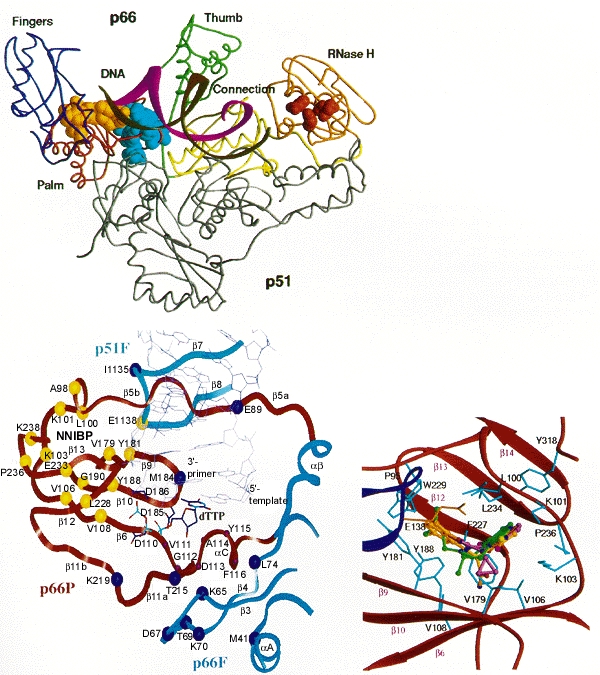
As noted above, an important problem limiting the therapeutic usefulness of nucleoside analogs is the selection in treated patients of viral variants resistant to inhibition by these compounds. Resistance has been associated with specific amino acid substitutions at a number of RT residues (Table 5; Fig. 11). Loss of HIV-1 susceptibility to AZT is correlated with substitutions at residues 41, 67, 70, 215, and/or 219 (Larder et al. 1989; Larder and Kemp 1989). Different combinations of substitutions result in different levels of resistance. It has been difficult to investigate the mechanism of resistance associated with the mutations at residues 41, 67, 70, 215, and 219 because most workers have found that purified RT with these changes is not resistant to AZTTP in vitro. It is unclear why this problem has been so difficult; in other cases, including AZT resistance caused by a mutation at position 151 (Shirasaka et al. 1995), purified HIV-1 RT mutants do show the expected and appropriate resistance in in vitro assays. Clinically significant resistance to ddI seems to be engendered primarily by substitutions at positions 65 and 74 (St. Clair et al. 1991; Gu et al. 1994; Zhang et al. 1994) and loss of susceptibility to ddC has been associated with substitutions at residues 65 and 69 (Fitzgibbon et al. 1992; Gu et al. 1992, 1994; Zhang et al. 1994). It is unclear how alterations at most of these RT positions affect viral susceptibility since, according to the available RT crystal structure, the substitutions associated with resistance are in positions that are relatively far from the polymerase active site (see Fig. 12B) (Jacobo-Molina et al. 1993). Most of the changes are in positions where RT interacts with the template-primer, and it has been proposed that it may be that the active site is influenced, and resistance engendered, by changes in enzyme–template-primer interactions that enable the enzyme to discriminate between normal dNTPs and the analogs (Boyer et al. 1994b; Tantillo et al. 1994). Substitution at residue 65 appears to affect adversely the ability of the enzyme to bind the nucleoside analog triphosphates (Zhang et al. 1994). In addition, amino acid substitution at residue 184 reduces the susceptibility of the virus to ddI, ddC, 3TC, and FTC (Gu et al. 1992; Boucher et al. 1993; Gao et al. 1993; Schinazi et al. 1993; Tisdale et al. 1993). In contrast to the other substitutions known to cause resistance to nucleoside analogs, this residue is located adjacent to two of the aspartate residues (185 and 186) that are part of the polymerase active site.
Table 5
Mutations in HIV-1 Associated with Drug Resistance.
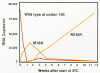
The seemingly facile acquisition of resistance to the nucleoside analogs has contributed to the limited clinical benefits that have been obtained when these inhibitors are used, particularly when they are used in monotherapy. Figure 13 demonstrates how rapidly resistant virus supplants the susceptible wild-type population in patients treated with 3TC monotherapy. This rapid emergence of resistance, which occurs after the administration of both nucleoside analogs and nonnucleoside inhibitors of RT (and, as will be discussed below, protease inhibitors as well), is a direct consequence of viral dynamics in the patients (for more details, see Chapter 11. Basically, since there are large numbers of viruses present in patients, and the error rate in copying the viral genome is fairly high, resistant variants preexist in a patient when therapy is initiated. Both the virus and virally infected cells turn over quite rapidly, which provides an opportunity for drug-resistant viral variants to emerge rapidly in patients. Nevertheless, the results of recent clinical trials suggest that various combinations of nucleoside analog RT inhibitors may have enhanced in vivo antiviral activity compared with what can be achieved when these same analogs are used in monotherapy. Some of this enhanced activity may reflect unfavorable interactions between RT amino acid substitutions that engender viral resistance to each of two inhibitors (discussed below).
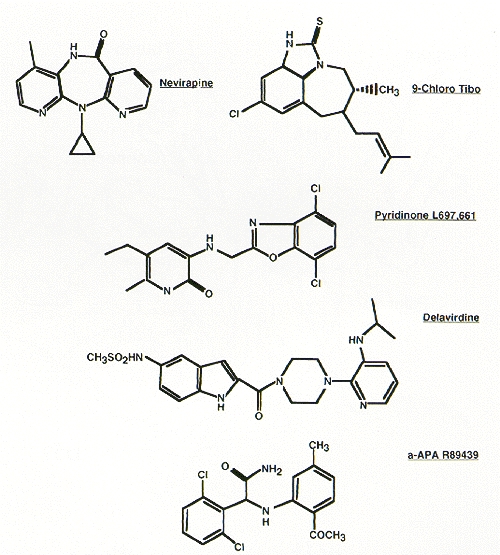
Nonnucleoside RT Inhibitors
The second major class of RT inhibitors is composed of compounds that are structurally diverse (see Fig. 14). However, all are noncompetitive (or mixed type) inhibitors of the enzyme and all are quite specific for HIV-1 RT. Some of the best studied members of this class include the thiobenzimidazolone derivatives R82150 and R82913 (Pauwels et al. 1990; White et al. 1991), the dipyridodiazepinone BI-RG-587 (Merluzzi et al. 1990), the pyridinone L-697,661 (Goldman et al. 1991), the bisheteroarylpiperazines (Dueweke et al. 1993b), the α-anilinophenyl acetamide derivatives (Pauwels et al. 1993), the PETT derivatives (Ahgren et al. 1995), and the benzoxazin-2-one, L-743,726 (Young et al. 1995). A large number of additional compounds that behave similarly have been described and are listed in Table 6, but they are not discussed here. Unlike the nucleoside analogs, the nonnucleoside inhibitors do not require metabolic activation. Clinical experience has shown that these compounds are generally well-tolerated, although some dose-limiting toxicities have been noted. However, in spite of this, in vivo antiviral effects have been transient because of the rapid selection of resistant viral variants (Davey et al. 1993; Saag et al. 1993; Richman et al. 1994). The replacement of the circulating viral population by resistant variants occurs within a few weeks after the start of therapy, which attests to the high rate of HIV-1 replication in vivo (Wei et al. 1995) (see also Chapter 11. A recent study with the nonnucleoside BI-RG-587 suggests that sufficiently high doses of the inhibitor may partially suppress the replication of some resistant viral mutants, thereby resulting in the prolongation of the antiviral effect in some treated patients (Havlir et al. 1995). However, the generality of this observation remains to be determined.
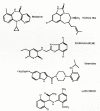
Figure 14
Structures of representative nonnucleoside HIV-1 RT inhibitors. Note that the structures are very different despite interacting at a common site in RT (Fig. 12).
Table 6
Partial Listing of Nonnucleoside HIV-1 Reverse Transcriptase Inhibitors.
It is possible to select, in cell culture, HIV-1 variants resistant to each of the reported nonnucleoside inhibitors. Resistance is generally the result of specific amino acid substitutions at RT residues 98 to 108 and/or 138, 179, 181, 188, 190, or 236 (see Table 5) (Cohen et al. 1991; Nunberg et al. 1991; Richman et al. 1991; Sardana et al. 1992; Bacolla et al. 1993; Balzarini et al. 1993a,b; Byrnes et al. 1993; Dueweke et al. 1993a; Mellors et al. 1993; Boyer et al. 1994a; Van Damme et al. 1994). As expected, these alterations affect the in vitro susceptibility of HIV-1 RT to the appropriate nonnucleosides. Each amino acid substitution exhibits notably different effects on resistance to the various inhibitors; however, there is considerable cross-resistance within the class. This suggests that these structurally diverse compounds occupy the same binding site on the RT, but each does so in a unique way. Crystallographic analyses of HIV-1 RT complexed with various nonnucleosides demonstrated that these inhibitors occupy a binding pocket that is located proximal to the polymerase active site (see Fig. 12C) (Kohlstaedt et al. 1992; Smerdon et al. 1994; Ding et al. 1995a,b; Ren et al. 1995a,b). What is somewhat surprising, considering the diverse nature of the compounds, is how similarly they bind RT; however, the specific drug/protein contacts do differ, which accounts for the patterns of resistance that are observed. Binding to this pocket adversely affects the rate of the chemical reaction catalyzed by the RT (Rittinger et al. 1995; Spence et al. 1995), and it may also inhibit the enzyme's flexibility. The amino acid residues that influence both viral and RT susceptibility to the nonnucleoside inhibitors make direct contributions to the structure of the binding pocket (see Fig. 12C) (Boyer et al. 1994a; Jonckheere et al. 1994; Tantillo et al. 1994); resistance involves residues that are directly involved in drug binding.
Other RT Inhibitors
Numerous other HIV-1 RT inhibitors, both nucleoside analogs and nonnucleoside compounds, have been described (for a comprehensive review, see De Clercq 1992). The clinical usefulness of many of these inhibitors is limited by either toxicity or lack of antiviral potency, or both. A compound that is generally active against many viral polymerases, including the HIV-1 RT, and has been used clinically in defined circumstances is phosphonoformate (PFA) (for review, see Oberg 1989). However, this compound is relatively nonselective and toxic. HIV-1 variants resistant to PFA have been reported (Tachedjian et al. 1995).
Combination Therapy with RT Inhibitors
Additive or synergistic in vitro antiviral activity is usually seen when a nucleoside analog is used together with a nonnucleoside RT inhibitor. This observation, along with the nonoverlapping resistance profiles of the two inhibitor classes, suggests the possibility of combination therapy. This concept is further supported by the observation that coexpression of substitutions at position 181, which is associated with resistance to nonnucleoside inhibitors, with any of the AZT-specific substitutions, results in viral variants that retain significant susceptibility to AZT (Larder 1992; Staszewski et al. 1995). Similarly, the ddI resistance-specific alteration at residue 74 mediates resensitization to AZT in certain contexts (St. Clair et al. 1991).
Clinically significant resistance to many of the RT inhibitors can, however, be mediated by multiple substitutions, and it usually is not possible to predict how various substitutions will functionally interact with each other (Byrnes et al. 1994). For instance, the nonnucleoside-specific substitutions at residue 103 or 106 will abrogate the AZT resensitizing effect of the change at position 74 when the latter is coexpressed with the AZT-specific substitution at position 215 (Emini et al. 1993; Larder et al. 1993). The simple conclusion based on an analysis of the resistance of such multiply substituted RTs is that the enzyme possesses extraordinary functional and structural flexibility. It has been possible to select in cell culture or to construct HIV-1 variants that are multiply resistant to a number of RT-directed combination therapies. Shafer et al. (1994) and Shirasaka et al. (1995) reported isolating, from patients receiving combination treatment with AZT plus ddI or ddC, mutant HIV-1 which expressed resistance to multiple dideoxynucleosides. The multiple resistance seems to be associated with the coexpression of a unique set of RT amino acid substitutions (Iversen et al. 1996).
There are, however, limitations on this flexibility which are discussed in more detail in a broader context at the end of this chapter. A specific example is seen in the inability to select rapidly for doubly resistant virus in patients undergoing combination therapy with AZT and 3TC (Kemp et al. 1994; Larder et al. 1995). The RT substitution at residue 184 normally associated with 3TC resistance was found to resensitize AZT-resistant virus to AZT (Tisdale et al. 1993). This suggests a functional incompatibility between 3TC- and AZT-associated substitutions that may make the selection for doubly resistant virus difficult. In turn, this may partially explain the sustained antiviral effect seen in patients treated with 3TC and AZT combination therapy (see above Nucleoside Analog Inhibitors).
Inhibition of the RNase H Activity of RT
As is true for all retroviral RTs, the RT of HIV-1 has an associated RNase H activity that is responsible for degrading the viral RNA template during the synthesis of the viral DNA. HIV-1 RT is a heterodimer; the RNase H domain is composed of the carboxy-terminal segment of the larger subunit, p66 (see Chapter 4 (Hansen et al. 1988; Prasad and Goff 1989; Hizi et al. 1990). Mutations that render RNase H inactive abolish viral infectivity, establishing RNase H as a suitable target for therapeutic intervention (Tisdale et al. 1991).
Little work has been reported on the development of specific inhibitors. Sulfated polyanions appear to be potent inhibitors of the RNase H of HIV-1 RT, but these compounds lack specificity and cannot be developed into orally available drugs (Moelling et al. 1989). The search for inhibitors is aided by the availability of a high-resolution crystal structure of the HIV-1 RNase H domain (Davies et al. 1991) and by a recombinant RNase H domain that is enzymatically active (Evans et al. 1991).
Proviral Integration
Following reverse transcription of the viral RNA genome, the resulting double-stranded linear viral DNA is integrated into the genome of the host cell (see Chapter 5. This event, mediated by the viral enzyme integrase (IN), involves two distinct steps. The first is a site-specific endonucleolytic removal of the 3′-terminal dinucleotide from the LTR sequences at each end of the provirus (Brown et al. 1989; Katzman et al. 1989; Roth et al. 1989; Sherman and Fyfe 1990). The second is a concerted strand-transfer reaction that joins the recessed 3′termini of the viral DNA to the 5′ends of target DNA (Fujiwara and Mizuuchi 1988; Brown et al. 1989; Roth et al. 1989; Katz et al. 1990).
The HIV-1 IN has an amino-terminal zinc finger that may be involved in specific recognition of the DNA substrate (Khan et al. 1991; Burke et al. 1992), an enzymatic core that is conserved among the retroviral INs (Khan et al. 1991; Engelman and Craigie 1992; Kulkosky et al. 1992; LaFemina et al. 1992), and a carboxy-terminal DNA-binding domain. Genetic alterations of the zinc finger motif reduce specific DNA cleavage activity of the enzyme (Engelman and Craigie 1992; LaFemina et al. 1992; Vincent et al. 1993; Hazuda et al. 1994b), whereas alteration of the catalytic core abolishes all enzymatic activity (Drelich et al. 1992; Engelman and Craigie 1992; LaFemina et al. 1992; van Gent et al. 1992; Vincent et al. 1993). Introduction of substitutions that inactivate IN into an HIV-1 provirus renders the resulting virus incapable of establishing productive infection of T-lymphoid cell lines or of primary cultures of macrophages and peripheral blood mononuclear cells (see LaFemina et al. 1992; Cannon et al. 1994; Wiskerchen and Muesing 1995). IN may not be an optimal therapeutic target both because of the stability of the preintegration complex (see Zack et al. 1992; Bukrinsky et al. 1993) and because a limited number of reactions is required to mediate the integration event. A successful in vivo inhibitor may require extraordinarily high enzyme-binding activity or may have to irreversibly disrupt the preintegration complex.
To date, several chemicals have been described that inhibit IN in vitro (Fesen et al. 1993; Mazumder et al. 1994; LaFemina et al. 1995). However, none of these compounds exhibit antiviral activity that can be specifically ascribed to inhibiting IN. An exception may be an oligonucleotide, T30177, which has anti-HIV-1 activity in cell culture that may be partially due to its ability to inhibit IN (Ojwang et al. 1995).
The lack of a previously studied cellular homolog of IN means that there is no precedent for the design of IN inhibitors. In vitro inhibitor assays for recombinant IN have relied primarily on radioactively labeled oligonucleotide substrates. Analysis involves fractionation by gel electrophoresis (for reviews, see Brown 1990; Goff 1992); however, microtiter plate-based assays that are more suitable for large-scale screening efforts have been developed (Hazuda et al. 1994a; Vink et al. 1994). The recent report of the IN catalytic core crystal structure provides a basis for structure-based drug design efforts that will complement screening (Dyda et al. 1994).
Virion Proteolytic Maturation
Immediately following budding of the retroviral particle at the host-cell plasma membrane, the viral protease cleaves the Gag and Gag-Pol polyproteins. These cleavages produce the mature viral core proteins and virus-specific, core-associated enzymes (see Chapters 2 and 7). The HIV-1 protease (PR) was originally identified on the basis of sequence similarity to the PRs of other retroviruses (Ratner et al. 1985). It was postulated that the enzyme was an aspartyl PR (Toh et al. 1985) and that, unlike similar mammalian enzymes, the viral PR was composed of two identical subunits (Pearl and Taylor 1987). Each subunit of the PR homodimer contributes one of the two aspartic acid residues at the active site. Mutagenic alteration of these residues results in an inactive enzyme (Kohl et al. 1988; LeGrice et al. 1988; Seelmeier et al. 1988).
The potential of the HIV-1 PR as a therapeutic target was established by mutagenesis of the codon for the active site aspartic acid residues in an infectious provirus (Kohl et al. 1988). Upon transfection, the mutant provirus directed the production of noninfectious immature viral particles that contained unprocessed viral Gag and Gag-Pol polyproteins.
The development of HIV-1 PR inhibitors relied heavily on previous work with aspartyl PRs such as renin and on a precise knowledge of the crystallographic structure of the enzyme (Lapatto et al. 1989; Navia et al. 1989; Wlodawer et al. 1989; Chen et al. 1994). Peptide cleavage studies using recombinant PR showed that the minimal substrate has seven amino acid residues. These residues extend from the P4 to P3′positions (with cleavage occurring at the amide bond between P1 and P1′) (Darke et al. 1988; Kotler et al. 1988; Lillehoj et al. 1988; see also Chapter 7. Most of the interactions of the enzyme and the substrate are stabilized by hydrogen bonds with the backbone of the substrate and by specific interactions with the amino acid side chains at the P3 and P3′positions (Miller et al. 1989; Erickson et al. 1990; Fitzgerald et al. 1990).
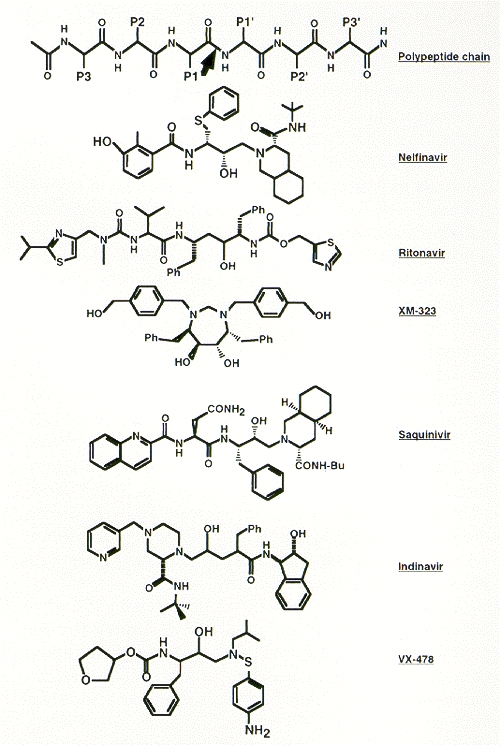
Numerous inhibitors of the HIV-1 PR have been described (for review, see Huff 1991). Figure 15 presents the structures of some inhibitors that have undergone clinical evaluation. Some of the first inhibitors that were synthesized incorporated hydroxyethylene or dihydroxyethylene isostere transition state analogs at the position where the enzyme would cleave the amide bond within a natural peptide substrate (Dreyer et al. 1989; Ashorn et al. 1990; McQuade et al. 1990; Meek et al. 1990; Tomaselli et al. 1990). Subsequently, smaller and highly potent compounds, incorporating the hydroxyethylene isostere motif, were synthesized (Lyle et al. 1991; Vacca et al. 1991; Thompson et al. 1992), eventually leading to the development of the HAPA (hydroxyaminopentane amide) inhibitors (Dorsey et al. 1994; Vacca et al. 1994). These compounds incorporate a basic amine into the hydroxyethylene isostere inhibitor backbone to enhance aqueous solubility and oral bioavailability. MK-639 (L-735,524), a HAPA compound, prevents spread of HIV-1 infection in cell culture at nanomolar concentrations and exhibits reasonable bioavailability when given orally to test animals using clinically acceptable formulations (Vacca et al. 1994).
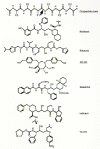
Figure 15
Structures of representative HIV-1 protease inhibitors. The top line shows a polypeptide backbone and the site of PR cleavage (arrow) for comparison.
An independent series of studies resulted in the synthesis of Ro 31-8959 (Roberts et al. 1990; Galpin et al. 1994). This compound incorporates an hydroxyethylamine isostere structure and exhibits antiviral activity in culture that is similar to MK-639. A third series of inhibitors was developed based on the enzyme's apparent symmetry; these are the C2 symmetry-based inhibitors (Erickson et al. 1990; Kempf et al. 1990, 1991; Otto et al. 1993b). A-77003 and A-80987 are examples of such compounds. In cell culture, these inhibitors are somewhat less potent than either MK-639 or Ro 31-8959. Recently, an inhibitor containing the novel (R)-(hydroxyethyl) urea isostere, SC-52151, was described (Bryant et al. 1995).
Computer-assisted design, based on the crystal structure of protease and of the structure-activity relationships of various C2-symmetric inhibitors, led to the development of nonpeptidyl PR inhibitors. The most potent of these described so far, XM-323, is active at nanomolar concentrations (Lam et al. 1994; Otto et al. 1993a). Reich et al. (1995) have described similar design studies using structural knowledge of cocrystallized inhibitor-PR complexes.
Clinical studies of several HIV-1 PR inhibitors have demonstrated that these compounds can profoundly suppress the levels of circulating virus (Collier et al. 1994; Deutsch et al. 1994; Massari et al. 1995). With certain compounds, the reduction in viral load was greater than 99%. However, in some patients, the antiviral effect appeared to be transient. As with the RT inhibitors, treatment with these drugs selects for resistant viral variants (Deutsch et al. 1994).
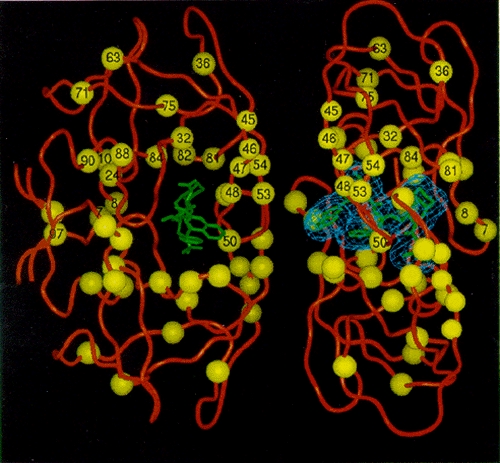
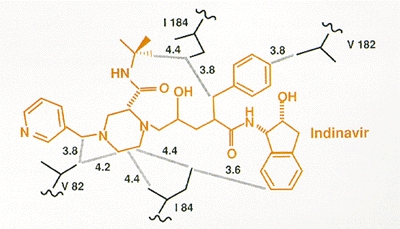
HIV-1 variants expressing reduced susceptibility to several PR inhibitors have been derived in cell culture (Craig et al. 1993; Dianzani et al. 1993; Otto et al. 1993b; Ho et al. 1994; Kaplan et al. 1994; Jacobsen et al. 1995; King et al. 1995; Markowitz et al. 1995; Partaledis et al. 1995; Patick et al. 1995, 1996; Tisdale et al. 1995). Isolation of these variants apparently requires a greater number of passages in culture in the presence of the drug than are needed to obtain resistant variants to the nonnucleoside RT inhibitors (see above). After 24 weeks of treatment with suboptimal doses of the PR inhibitor MK-639, viral variants were isolated from patients that showed a significant decrease in susceptibility to the compound (Deutsch et al. 1994; Mellors et al. 1995). Genetic analysis of these clinically derived mutants demonstrated that resistance was due to amino acid substitutions at a number of residues in PR, including 10, 20, 24, 46, 54, 63, 64, 71, 82, 84, and 90 (Fig. 16). However, analysis of clinical isolates and of viral variants produced by site-directed mutagenesis showed that the expression of single or double amino acid substitutions was not sufficient to induce resistance. Rather, combinations of three or more amino acid alterations were required (Condra et al. 1995, 1996). However, the substitution patterns appeared to vary among the resistant clinical isolates. In general, higher levels of resistance were associated with the coexpression of greater numbers of substitutions.
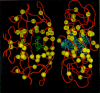
Selection for amino acid substitutions in PR often occurred prior to the isolation of measurably (by cell culture assay) resistant virus. In addition, alterations continued to accumulate even after resistant variants had been selected. It may be that the amino acid substitutions selected initially provide a small selective advantage in the presence of inhibitor, although these small effects are not obvious in laboratory assays of viral susceptibility. Measurable resistance is often associated with changes in PR residues, for example, at positions 82 and 84, that are within the enzyme active site and that interact directly with the inhibitor (Fig. 17). Selection for additional alterations in PR, particularly for alterations that are not within the active site, may reflect the selection of compensatory changes that help the enzyme accommodate the active site changes without significant loss of viral replicative capacity (discussed at the end of the chapter).
The probability of selection for resistant viral variants during the first 24 weeks of therapy with MK-639 appeared to depend on the dose of the inhibitor. When higher initial doses were used, a larger number of patients exhibited sustained profound decreases (>99%) in circulating viral levels than when the initial doses were lower (Massari et al. 1995; Mellors et al. 1995). The apparent ability to forestall the emergence of resistant viruses when higher doses were used may reflect the need for selection of a complex array of mutations to produce resistance to the inhibitor. The higher dose may partially inhibit the replication of the initial variants, which may slow the selection of the full suite of the amino acid substitutions needed to give high-level resistance. It seems that variants that contain the amino acid substitutions associated with resistance to PR inhibitors are poorly represented in the wild-type viral populations, suggesting that such mutant viruses are at a disadvantage relative to the wild type (Coffin 1995).
At least two studies have demonstrated that the selection for virus resistant to one PR inhibitor can produce variants that are cross-resistant to other, structurally diverse PR inhibitors (Condra et al. 1995; Tisdale et al. 1995). The genetic basis for cross-resistance appears to be complex, and the exact patterns of cross-resistance among the inhibitors vary considerably. However, sufficient overlap exists among the amino acid substitutions in PR that contribute to resistance to suggest that previous therapy with one inhibitor can adversely influence the benefit of follow-up treatment with a second inhibitor. As discussed above, treating patients with a suboptimal dose of an inhibitor negates the expected antiviral effect when the dose is raised to the optimal level (Mellors et al. 1995). This reflects the preselection, at the lower dose, of viral variants that are partially resistant to the higher dose. A similar phenomenon may occur when two different inhibitors are used sequentially.
Virion Maturation
Several other approaches intended to inhibit lentiviral maturation have been described. The first involves the constitutive expression, within susceptible cells, of mutant viral Gag proteins that interfere with HIV-1 virion core assembly (Trono et al. 1989). These modified cells are incapable of supporting production of infectious virions even if a provirus is introduced into them by transfection. This is an example of the large class of dominant-negative mutants and the approach is an illustration of the concept of “intracellular immunization” (Baltimore 1988). Another version of this strategy involved the derivation of a cell line that expressed a mutated CD4 receptor molecule which contains a signal for retention in the endoplasmic reticulum (Buonocore and Rose 1990). When these cells were infected, the variant receptor bound to the viral envelope glycoprotein within the cell, preventing transport of the Env protein to the surface. A dominant mutant of the HIV-1 gp41 transmembrane glycoprotein that interferes with the production of infectious virions has also been described (Freed et al. 1992). Similar intracellular immunization schemes have been described using the HIV-1 regulatory proteins as targets (see below). The practical obstacles facing the therapeutic implementation of such approaches are significant and are discussed below (see below Ribozymes).
Finally, it has been shown that the HIV-1 Gag and Gag-Pol polyproteins are targeted to the plasma membrane of the infected cell by a covalently linked myristic acid moiety at the amino terminus of the polyprotein (Bryant and Ratner 1990; Pal et al. 1990). Accordingly, interference with myristylation prevents viral replication (Bryant et al. 1989). However, given the ubiquitous role of myristylation in uninfected cells, this target may not have sufficient selectivity to be used for pharmacologic intervention.
Viral Regulatory Proteins
The replicative cycle of lentiviruses is complex, and its regulation involves both cellular and viral factors that control the production, processing, and intracellular transport of viral RNAs (see Chapters 6 and 11). The cellular factors are not discussed here because of the obvious lack of uniqueness required for a therapeutic target. In contrast, lentiviruses express two unique regulatory proteins, Tat and Rev, that control the production and cytoplasmic localization of viral RNA (Cullen 1992; see also Chapter 6.
A number of approaches intended to control the function of Tat and Rev have been described. This section considers chemical inhibitors and dominant-negative mutants; antisense oligonucleotides and ribozymes are described below (see Antisense Inhibitors and Ribozymes).
Tat
The viral Tat protein binds to a region of specific secondary structure located near the 5′end of the viral RNA. Binding of Tat to this structure, termed TAR, results in enhanced viral RNA production. This is apparently accomplished because the interaction prevents premature termination of transcription. In addition to interacting with TAR, Tat binds to cellular proteins that may be required for its activity. Both of these binding interactions can be used to derive dominant-negative mutants (see above). Two such mutants have been described (Pearson et al. 1990; Modesti et al. 1991). The mechanism of action of the mutants is not clear, but it does not involve a block in RNA binding. Rather, expression of the mutant protein may result in the formation of inactive Tat complexes or may sequester required Tat-binding cellular proteins.
Other schemes involve the overexpression in susceptible cells of decoy TAR RNA targets (Sullenger et al. 1991) or the intracellular expression of anti-Tat single-chain antibodies (Mhashilkar et al. 1995). In these examples of intracellular immunization, the modified cells were unable to produce infectious virus.
Finally, a chemical compound that appears to inhibit HIV-1 Tat has been described (Hsu et al. 1991). This compound, Ro 5-3335, is a benzodiazepine derivative that reduces the levels of HIV-1 RNA in both acutely and chronically infected cells. The exact molecular target is undefined, but both Tat and TAR are required. However, clinical studies of this compound and its analogs (e.g., Ro 24-7429) suggest that these inhibitors have insufficient potency for significant in vivo antiviral activity (Haubrich et al. 1995).
Rev
The HIV-1 Rev protein binds to a specific region of the viral RNA called the Rev response element (RRE). Rev binding to RRE increases the amount of singly spliced and unspliced viral mRNAs in the cytoplasm. These RNAs encode the viral structural proteins (see Cullen 1992; Chapters 6 and 7). The Rev protein has a number of functional domains, including a domain required for a multimerization step that Rev must undergo for proper activity. Hence, like Tat, Rev is an appropriate target for dominant-negative mutants. Such mutants have been developed and were shown to inhibit HIV-1 replication in target cells (Malim et al. 1989, 1992; Mermer et al. 1990; Bevec et al. 1992). The mutant protein does not appear to compete with the wild-type Rev for binding to the target RNA. Rather, mutant Rev apparently interacts with wild-type Rev, which could result in the formation of nonfunctional complexes (Hope et al. 1992). A dominant-negative Rev mutant that prevents nucleolar localization of multimeric Rev has also been described (Kubota et al. 1992). In addition, intracellular expression of an anti-Rev single-chain antibody was shown to inhibit HIV-1 replication in cell culture (Duan et al. 1994). Viral replication has also been inhibited in cell culture by the overexpression of RRE fragments that bind Rev (Lee et al. 1992; Lee et al. 1994; Bevec et al. 1994). Specific chemical inhibitors of Rev activity have not yet been described.
Antisense Inhibitors and Ribozymes
This section focuses on two novel approaches that can be directed to multiple viral targets. Both approaches attempt to take advantage of the specific interactions between complementary nucleic acid molecules.
Antisense Oligonucleotides
Antisense oligodeoxynucleotides have been prepared that are complementary to a variety of HIV-1 RNA sequences. A number of these oligonucleotides were demonstrated to exhibit antiviral activity in cell culture (Zamecnik et al. 1986; Matsukura et al. 1987, 1989; Agrawal et al. 1988, 1989; Goodchild et al. 1988; Lisziewicz et al. 1994). The molecular mechanism of inhibition has not been precisely defined for many of the inhibitory oligonucleotides. It seems that, in certain cases, inhibition is dependent on sequence-specific interactions (Matsukura et al. 1989). Inhibition may result from stearic hindrance or from RNase H-mediated degradation of viral RNA complexed with a complementary oligodeoxynucleotide. In other cases, viral inhibition is obviously due to undefined nonspecific activities of the oligonucleotide (Matsukura et al. 1987; Agrawal et al. 1988, 1989; Kinchington et al. 1992). In these situations, random oligonucleotides and homooligomers displayed antiviral effects in cell culture.
Antisense oligonucleotides (both DNA and RNA) with putative antiviral activity have been targeted to the 5′leader sequences of the viral gag mRNA (Sczakiel and Pawlita 1991), Tat mRNA (Joshi et al. 1991), the RRE and rev gene (Chin 1992; Lisziewicz et al. 1992), the TAR sequence (Vickers et al. 1991), the Tat RNA-coding region (Lo et al. 1992), the splice acceptor site of the tat gene (Daum et al. 1992), and the U5 region of the viral genome and a region proximal to the primer-binding site (Bordier et al. 1995). HIV-1 replication in cell culture has also been inhibited by introducing into cells a vector expressing an antisense RNA molecule directed against the RT-coding region (Meyer et al. 1993).
Antisense oligonucleotide therapy faces significant practical concerns. The most important of these are delivery and uptake by target cells and the stability of the oligonucleotide upon systemic administration. Considerable progress has been made toward the large-scale synthesis of oligonucleotide chemical derivatives that can penetrate cells and are stable to the nucleases found in serum and target cells. Examples include the phosphorthioate oligonucleotide analogs (for review, see Stein and Cheng 1993). Such analogs are currently undergoing initial clinical evaluation.
Ribozymes
Ribozymes are catalytic RNA molecules that cleave RNA substrates. Ribozyme specificity is dependent on complementary RNA-RNA interactions (for review, see Cech and Bass 1986). Two types of ribozymes, hammerhead and hairpin, have been described. Each has a structurally distinct catalytic center. Hammerhead ribozymes were initially demonstrated to mediate the cleavage of several HIV-1 RNA transcripts (Chang et al. 1990). These ribozymes, when stably expressed in susceptible human cells, were found to inhibit HIV-1 replication (Sarver et al. 1990). To date, hammerhead ribozymes have been described that target the HIV-1 vif RNA sequences (Lorentzen et al. 1991), the HIV-1 5′-leader RNA sequence (Weerasinghe et al. 1991; Homann et al. 1993), the integrase-coding RNA sequence (Sioud and Drlica 1991), the tat gene (Lo et al. 1992), the HIV-1 U5 RNA sequence (Dropulic et al. 1992), the envelope glycoprotein-coding RNA sequence (Chen et al. 1992), the HIV-1 LTR RNA (Heidenreich and Eckstein 1992), and the viral RNA packaging sequence (Sun et al. 1994). In addition, a hairpin ribozyme, which is specific for the viral leader sequence, has been shown to inhibit HIV-1 infection in cells that express the ribozyme (Ojwang et al. 1992).
The obstacles facing the use of ribozymes in practical therapeutic approaches, or for that matter intracellular immunization or targeted antisense therapy, are substantial. As discussed in Chapter 9 a major obstacle in gene therapy is the efficient delivery of the vector; there are also significant issues involving the immunological reaction of the host. It may be possible to introduce expression vectors for ribozymes or for dominant-negative proteins by ex vivo transfection of bone marrow cells. The transfected, and presumably protected, cells can then be implanted into depopulated marrow. The efficiency of this method is relatively poor and thus many of the target cells will not be transfected. Given the high rate of HIV-1 replication and turnover in vivo, such partial putative protection of target cells may not manifest itself as a significant antiviral effect. Alternatively, retroviral or other viral vectors may be used to transduce appropriate genes into cells both ex vivo and in vivo. One method involves the use of adeno-associated virus encoding antisense RNA (Chatterjee et al. 1992). However, the expression of HIV-1 proteins (or for that matter, vector proteins) is likely to provoke the immune system to eliminate the modified cells.
Other Potential Approaches
As further understanding of the biological functions associated with various HIV-1 proteins is obtained, additional targets for chemotherapeutic intervention may well emerge. For example, there have been a number of reports that the nonstructural protein Nef has a role in the production of viral particles that will efficiently synthesize proviral DNA (Chowers et al. 1994; Aiken and Trono 1995; Schwartz et al. 1995). How Nef causes this phenotypic effect is unclear, but Saksela et al. (1995) have suggested that Nef mediates the phosphorylation of viral structural proteins by cellular kinases. If so, this pathway may be accessible to pharmacologic intervention. Study of the biochemical activities of other important HIV-1 regulatory proteins, such as Vpr and Vif, may also provide novel targets.
A novel chemotherapeutic approach described by Rosenwirth et al. (1994) demonstrated that a nonimmunosuppressive variant of cyclosporin A could inhibit HIV-1 infection in cell culture. The target of the inhibitor appears to be cyclophilin A, which is incorporated into HIV-1 virions (Franke et al. 1994; Thali et al. 1994). The actual function of cyclophilin A in the viral life cycle is probably to aid in the proper folding of Gag; this interaction might serve as the basis for the development of novel agents with antiviral effects.
A final example of a novel target is the zinc “finger” present in the nucleocapsid protein. This structure is apparently unique among protein zinc fingers, thereby raising hopes that compounds may be found that can specifically mediate its oxidation. Rice et al. (1995) and Tummino et al. (1996) reported a series of compounds intended to disrupt the zinc finger in the nucleocapsid; these compounds exhibited anti-HIV-1 activity in cell culture. However, the selectivity of the compounds and specificity of the target remain to be established.
Clinical Evaluation of Novel Anti-HIV Therapies
As with all new therapeutic agents, the clinical evaluation of anti-HIV drugs is divided into a series of (more or less) sequential phases. Phase I studies are typically performed in healthy volunteers following extensive safety, toxicity, genotoxicity, and pharmacologic studies done both in cell culture in vitro and in animals. Phase I evaluations are short-term and are designed to assess the pharmacology and toxicity of the test compound in humans. If the data and observations are acceptable, phase II studies are performed in patient populations for whom the drug is intended. These studies are also limited in size and are intended primarily for determining dosages, assessing tolerability, and, importantly, assessing the in vivo activity of the compound. The latter involves measuring the effect of the drug on a measurable marker of the target.
Once the appropriate dose has been selected on the basis of such trials, phase III studies are performed. These trials, carried out in larger numbers of patients, are designed to provide additional data on how well the drug is tolerated and to provide evidence of clinical benefit. The combined data from the three phases of evaluation are used to support the licensure of the drug by regulatory authorities. Clinical evaluations following licensure (phase IV) are performed to obtain additional tolerability and efficacy data or, in some cases, to assess the benefit(s) of using the drug for additional clinical situations.
The fatal nature of HIV-1 infection imposes an urgency on the clinical study of novel anti-HIV agents. In the United States, this urgency has led to the formulation of rules that would permit the “accelerated” approval of new drugs based on changes, mediated by these agents, in patient CD4 cell count and blood viral levels. It is becoming increasingly clear that these two parameters are predictive of progression to HIV-related clinical disease (Hammer 1996; Mellors 1996). This is especially true of changes in the viral load. As a result, phase II studies are designed to provide antiviral activity data to support accelerated licensure applications. Such approval, if granted, is provisional pending data from a phase III study which demonstrates clinical benefit defined as significant increases in time to disease progression or death.
The design and interpretation of clinical trials for anti-HIV therapies are complicated both by the prolonged course of the disease and by the fact that the antiviral effects of drugs can be altered by selection of a drug-resistant virus. In patient populations with advanced disease (characterized by low CD4 cell counts and high viral loads) in whom disease progression can occur over a relatively short time frame (months), the association between transient antiviral effects and clinical benefit can be established. However, in less-advanced patient populations in whom disease progression occurs over a much longer time period (years), it is much more difficult (if not impossible) to observe clinical benefit following a relatively brief period of antiviral treatment. Many of the phase III studies reported to date have been in advanced patient populations, and the positive clinical effects seen in such patients with monotherapy with relatively weak antiviral agents such as AZT have not been obtained when less-advanced patients are treated. Nevertheless, with the advent of more potent therapeutic agents that can mediate sustained antiviral effects (in particular with the new combination therapies), it may be possible to demonstrate clinical benefit in patients with relatively high CD4 cell counts and lower viral burden. Indeed, if anti-HIV chemotherapy is to be used for the chronic management of this persistent infection, then therapeutic regimens must be designed that will manifest positive clinical benefits in such long-term (years) phase III trials.
Strategies for Anti-HIV Therapy
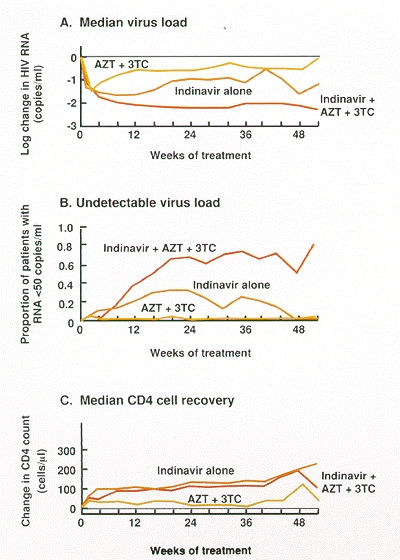
The availability of drugs that can profoundly affect the replication of HIV-1 in patients, combined with the facile development of resistance to these drugs, raises important questions about choosing the most effective therapeutic regimens. There is no currently available drug that is an effective inhibitor of HIV replication to which the virus does not rapidly develop resistance when it is used alone (i.e., in monotherapy). Effective and durable suppression of viral replication requires the use of drug combinations. Combination chemotherapy is not a novel idea: It has long been important in the treatment of certain bacterial infections (notably tuberculosis) as well as cancer. Although a number of combinations of inhibitors have been tried, the most effective of the therapies involves three drugs: two RT inhibitors (usually two nucleoside analogs, such as AZT plus 3TC) and a protease inhibitor (Gulick et al. 1997; Hammer et al. 1997). The combination of two nucleosides, although more beneficial than AZT alone (Eron et al. 1995), does not seem to be sufficient to block the replication of the virus to a degree that prevents the emergence of resistant strains. Even if replication of wild-type virus is completely blocked, preexisting mutants, which can accumulate in the absence of drug (Coffin 1995; Najera et al. 1995), can grow out rapidly after initiation of therapy, leading to failure within a few weeks or months (see Fig. 13) (Schuurman et al. 1995). Protease inhibitors, when used alone, give a longer period of suppression, but resistant mutants eventually appear in the large majority of HIV-infected individuals.
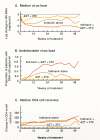
In contrast, recent clinical trials have shown that reductions in virus load and increases in CD4 cells can be sustained for at least 1 year using a combination of indinavir, AZT, and 3TC (Gulick et al. 1997; Hammer et al 1997). Indeed, more than two thirds of patients treated with this combination lacked detectable virus (50 copies of RNA per milliliter) in their blood 1 year after the initiation of treatment, whereas no patient receiving either indinavir alone or AZT plus 3TC showed a durable response (Fig. 18). These studies imply that viral replication can be controlled, but only with triple combination therapy. The current best regimen is expensive, and it is not yet known whether drug-resistant virus will eventually appear after a long period of suppression. Nevertheless, in the relatively short time since its introduction, the application of the triple combination therapy has already had a significant effect on HIV-associated morbidity and mortality in the United States (CDC 1997).
The rapid failure of most single- and two-drug combinations also means that if a patient carries a strain of virus that is resistant to one of the drugs in the three-drug regimen, then the three-drug therapy is effectively reduced to a two-drug therapy. This situation often arises in patients who have previously been treated with suboptimal regimes. Treating patients sequentially in this way will almost certainly fail to achieve long-term control of the infection, and it will eliminate drugs from a relatively limited armamentarium and reduce the effectiveness of future treatment. It is therefore particularly important not only to initiate therapy with a potent (three-drug) regimen, but also, if possible, to guard against the possibility that a patient is infected with a viral strain already resistant to one or more of the drugs to be used.
There is also the question of when to initiate therapy. In the past, when AZT was the only available option, drug therapy was usually initiated relatively late in infection, at the stage where the patient had begun to display symptoms of frank AIDS (see Chapter 11. Although combination therapy has caused dramatic improvements in many such patients, the current thinking is that it is better to initiate therapy much earlier in the course of infection. Not only will this strategy minimize the accumulation of irreversible damage to the immune system, but it should also give the best chance for minimizing the accumulation of mutations that can give rise to resistance. The proposal to treat patients early and aggressively with combinations of drugs (usually, but not necessarily, protease inhibitors combined with RT inhibitors) has recently become the recommendation of international committees for standard therapy of HIV infection (Carpenter et al. 1997).
The prolonged and substantial suppression observed in combination therapy (see Fig. 18) has raised the possibility that eradication of virus from an infected individual might be possible. It is still too early to tell whether eradication can be achieved. Although the vast majority of infected cells disappear rapidly after initiation of treatment, there are, in all patients, smaller reservoirs of infected cells that have much longer life spans (Perelson et al. 1997). Eliminating the virus requires not only blocking active viral replication completely, but also doing so for a sufficient time that all of the long-lived cellular reservoirs have a chance to die out. Various estimates of the necessary therapeutic period have been put forward, depending greatly on both the lifetime and the size of the longest-lived cell population. Because neither of these values is presently known, estimates range widely from few years to a few decades. In considering the possibility of eradication, it is important to keep in mind that there is presently no way to be sure that viral replication is completely suppressed. If this cannot be achieved, then the appearance of HIV mutants resistant to any antiviral strategy, although it may be long delayed, will be inevitable, and even the best treatments will ultimately fail.
Publication Details
Copyright
Publisher
Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY)
NLM Citation
Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997. Pharmacologic Approaches.