Summary
Clinical characteristics.
PIK3CA-related overgrowth spectrum (PROS) encompasses a range of clinical findings in which the core features are congenital or early-childhood onset of segmental/focal overgrowth with or without cellular dysplasia. Prior to the identification of PIK3CA as the causative gene, PROS was separated into distinct clinical syndromes based on the tissues and/or organs involved (e.g., MCAP [megalencephaly-capillary malformation] syndrome and CLOVES [congenital lipomatous asymmetric overgrowth of the trunk, lymphatic, capillary, venous, and combined-type vascular malformations, epidermal nevi, skeletal and spinal anomalies] syndrome). The predominant areas of overgrowth include the brain, limbs (including fingers and toes), trunk (including abdomen and chest), and face, all usually in an asymmetric distribution. Generalized brain overgrowth may be accompanied by secondary overgrowth of specific brain structures resulting in ventriculomegaly, a markedly thick corpus callosum, and cerebellar tonsillar ectopia with crowding of the posterior fossa. Vascular malformations may include capillary, venous, and less frequently, arterial or mixed (capillary-lymphatic-venous or arteriovenous) malformations. Lymphatic malformations may be in various locations (internal and/or external) and can cause various clinical issues, including swelling, pain, and occasionally localized bleeding secondary to trauma. Lipomatous overgrowth may occur ipsilateral or contralateral to a vascular malformation, if present. The degree of intellectual disability appears to be mostly related to the presence and severity of seizures, cortical dysplasia (e.g., polymicrogyria), and hydrocephalus. Many children have feeding difficulties that are often multifactorial in nature. Endocrine issues affect a small number of individuals and most commonly include hypoglycemia (largely hypoinsulinemic hypoketotic hypoglycemia), hypothyroidism, and growth hormone deficiency.
Diagnosis/testing.
The diagnosis of PROS is established in a proband with suggestive findings and a heterozygous mosaic (or rarely, constitutional) activating pathogenic variant in PIK3CA. Sequence analysis of DNA derived from clinically affected tissue samples ‒ preferably from freshly obtained dermal biopsy overlying an affected area, from surgical excision of the overgrown tissue, or from uncultured tissues (such as skin fibroblasts or other tissues) ‒ should be prioritized for genetic testing. Targeted capture of the entire PIK3CA coding region followed by next-generation sequencing at very deep coverage is recommended for somatic variant detection, as it allows for detection of very low levels of mosaicism throughout the gene.
Management.
Targeted therapy: Alpelisib (VIJOICE®) 50 mg orally with food once a day (at about the same time every day) for those between age two years and <18 years with PROS. In those age six years or older, the dose may be increased to 125 mg once a day after 24 weeks. A starting dose of 250 mg orally with food once a day (at about the same time every day) has been approved for those age ≥18 years. Alpelisib has been approved specifically for the reduction of overgrowth, vascular lesions, and other functional complications. To date, it is unknown whether this drug has any efficacy in treating the neurologic manifestations of PROS (as, e.g., in MCAP syndrome).
Supportive care: Significant or lipomatous segmental overgrowth may require debulking; scoliosis and leg-length discrepancy may require orthopedic care and surgical intervention. Neurologic complications (e.g., obstructive hydrocephalus, increased intracranial pressure, progressive and/or symptomatic cerebellar tonsillar ectopia or Chiari malformation, and epilepsy in those with brain overgrowth/malformations) may warrant neurosurgical intervention. Depending on the type of vascular malformations, sclerotherapy, laser therapy, or oral medications such as sirolimus may be used. Similarly, lymphatic malformations may be treated through oral medications or careful surgical debulking, preferably by a vascular anomalies team. For those with pain, evaluation for the source of pain and treatment of the underlying cause is recommended. For those with growth hormone deficiency, evaluation of the hypothalamic-pituitary-adrenal axis is warranted; a trial of growth hormone therapy may be considered with careful monitoring of linear growth and overgrowth. Severe persistent hypoglycemia has been reported, and requires evaluation and ongoing treatment, which can include cornstarch administration. Routine treatment of the following, when present, is indicated: cardiac and renal abnormalities; intellectual disabilities and behavior issues; polydactyly and foot deformities; coagulopathy or thrombosis; Wilms tumor; and hypothyroidism.
Surveillance: At each visit: measurement of growth parameters including head circumference, length of arms, hands, legs, and feet; assess for new neurologic manifestations (seizures, changes in tone, and other signs/symptoms of Chiari malformation); monitor developmental progress and behavior; assess motor skills; clinical assessment for scoliosis and abdominal examination for organomegaly and/or abdominal masses. Serial head MRI imagining is recommended, with frequency based on the severity of findings on initial assessment and the degree of brain maturation. For those with CNS overgrowth or dysplasia, brain MRI every six months until age two years and then annually until age eight years to monitor specifically for progressive hydrocephalus and Chiari malformation. As clinically indicated: clinical assessment and monitoring of any vascular and/or lymphatic malformations; radiographs of the limbs in those with overgrowth of a limb or portion of a limb; ultrasound or MRI follow up in those with truncal overgrowth; spinal MRI in those with scoliosis or deformities that affect the spine; blood glucose monitoring and evaluation of the hypothalamic-pituitary-adrenal axis for those with persistent hypoglycemia, particularly if they require ongoing treatment for hypoglycemia. Hematology consultation with recommendations for assessment for thrombosis and coagulopathy risk after any surgical intervention, particularly in those with the CLOVES phenotype and/or those with vascular malformations. Consideration of renal ultrasound every three months until age eight years (tumor screening for Wilms tumor is controversial).
Genetic counseling.
PROS disorders are not known to be inherited, as most identified pathogenic variants are somatic (mosaic). No confirmed vertical transmission or sib recurrence has been reported to date. The risk to sibs of a proband with somatic mosaicism for a pathogenic variant in PIK3CA would be expected to be the same as in the general population. All but a few affected individuals with PROS have had somatic mosaicism for a PIK3CA pathogenic variant, suggesting that mutation occurred post fertilization in one cell of the multicellular embryo. Therefore, the risk for transmission to offspring is expected to be less than 50%.
GeneReview Scope
Table
Megalencephaly-capillary malformation (MCAP) syndrome Dysplastic megalencephaly (DMEG), hemimegalencephaly (HMEG) and focal cortical dysplasia (FCD)
Diagnosis
PIK3CA-related overgrowth spectrum (PROS) encompasses a range of clinical findings in which the core features are congenital or early-childhood onset of segmental/focal overgrowth with or without cellular dysplasia in the absence of a family history of similarly affected individuals (i.e., single occurrence in a family). Prior to the identification of PIK3CA as the causative gene, PROS was separated into distinct clinical syndromes based on the tissues and/or organs involved (see GeneReview Scope).
Suggestive Findings
PROS should be considered in individuals with the following clinical, brain MRI, and family history findings [Keppler-Noreuil et al 2015, Mirzaa et al 2016, Kuentz et al 2017].
Clinical features
- Overgrowth of any of a wide variety of tissues including (but not limited to) brain, adipose, vascular, muscle, skeletal, nerve
- Vascular malformations including (but not limited to) capillary, venous, arteriovenous, or mixed malformations
- Lymphatic malformations
- Cutaneous findings including epidermal nevi and hyperpigmented macules
- Single or multiple digital anomalies of the hands or feet (e.g., macrodactyly, syndactyly, polydactyly, sandal-toe gap)
- Kidney malformations
- Benign tumors, with the exceptions of Wilms tumor and nephroblastomatosis (i.e., diffuse or multifocal clusters of persistent embryonal cells)
Brain MRI findings. Focal brain overgrowth (with or without cortical dysplasia) including:
- Hemimegalencephaly (HMEG)
- Focal cortical dysplasia (FCD)
- Dysplastic megalencephaly (DMEG)
Family history. Because PIK3CA is typically caused by a de novo mosaic pathogenic variant, most probands represent a simplex case (i.e., a single occurrence in a family). Rarely, the family history may be consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations).
Establishing the Diagnosis
The diagnosis of PROS is established in a proband with suggestive findings and a heterozygous mosaic (or rarely, constitutional) activating pathogenic variant in PIK3CA [Keppler-Noreuil et al 2015] (see Table 1 and Molecular Genetics).
Molecular diagnosis. Molecular genetic testing approaches typically include use of targeted testing (single-gene testing or multigene panel), although comprehensive genomic testing (exome sequencing, genome sequencing) is available. Because the majority of reported PIK3CA pathogenic variants are postzygotic (and thus mosaic), more than one tissue (excluding blood in most cases) may need to be tested:
- Experience suggests that sequence analysis of DNA derived from clinically affected tissue samples ‒ preferably from freshly obtained dermal biopsy overlying an affected area, from surgical excision of the overgrown tissue, or from uncultured tissues (e.g., skin fibroblasts or other tissues) ‒ should be prioritized for genetic testing.
- The level of mosaicism for an activating variant in affected tissues or cultured cells is extremely variable [Keppler-Noreuil et al 2014, Kuentz et al 2017].
- Testing of blood or DNA isolated from blood is not recommended based on current technologies, as PIK3CA pathogenic variants have not been identified in blood except in two of 24 individuals with megalencephaly-capillary malformation (MCAP) syndrome, who had an apparently de novo germline pathogenic variant in PIK3CA [Rivière et al 2012].
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of PROS has not been considered may be diagnosed using genomic testing (see Option 2), if an appropriate sample is used.
Option 1
When the phenotypic findings suggest a diagnosis of PROS, molecular genetic testing approaches can include single gene testing or use of a multigene panel that includes PIK3CA.
- Single-gene testing. Sequence analysis of PIK3CA may be performed on an appropriate sample (see above) and can detect missense variants (see Molecular Genetics for a summary of appropriate laboratory techniques to detect low levels of mosaicism).Note: (1) The pathogenic variants observed in PROS have all been associated with gain of function; thus, gene-targeted deletion/duplication analysis is not recommended. (2) Failure to detect an activating PIK3CA pathogenic variant does not exclude a diagnosis of PROS in individuals with suggestive findings, given that a low level of mosaicism is observed in many affected individuals [Kurek et al 2012, Lee et al 2012, Lindhurst et al 2012, Jansen et al 2015, Mirzaa et al 2016].
- A multigene panel that includes PIK3CA and other genes of interest (see Differential Diagnosis) on an appropriate sample (see above) may be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the diagnosis of PROS has not been considered because an individual has atypical phenotypic features, genomic testing on an appropriate sample (see above) may be considered.
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in PIK3CA-Related Overgrowth Spectrum
Clinical Characteristics
Clinical Description
PIK3CA-related overgrowth spectrum (PROS) includes overgrowth of a broad range of tissues that may or may not be accompanied by cellular dysplasia. Prior to the understanding of the molecular nature of PROS, a number of distinct but overlapping phenotypes were clinically described and given names (see GeneReview Scope).
In general, PROS can be divided into an isolated form (when a person has a focal lesion that affects only one tissue or body part; see Table 2) and a syndromic form (i.e., overgrowth plus at least two other features in two systems; see Table 3). A targeted therapy aimed at inhibiting PI3K-related pathway overgrowth has been approved by the FDA (see Table 6).
Table 2.
Selected Isolated PIK3CA-Related Overgrowth Phenotypes by Affected Organ or Tissue
Table 3.
Selected Syndromic PIK3CA-Related Overgrowth Phenotypes
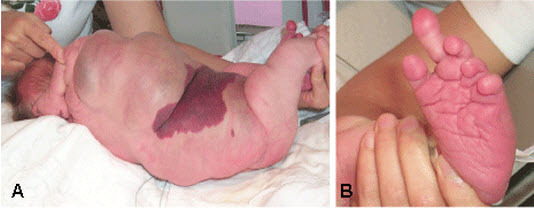
Figure 1.
Features of CLOVES syndrome in a child with (A) a large lipomatous truncal mass that extends into the surrounding tissues and an overlying capillary malformation and (B) macrodactyly of the left foot
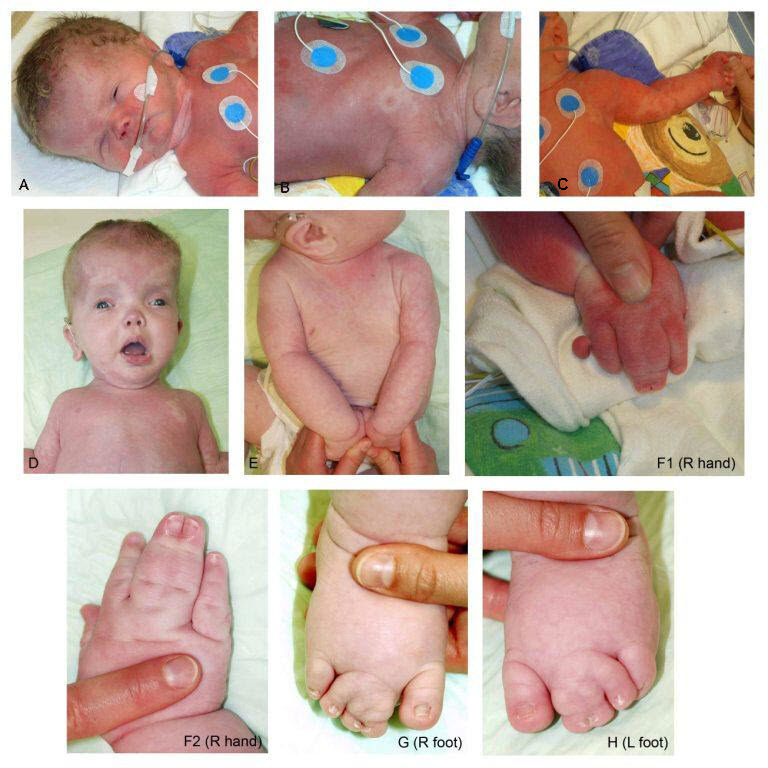
Figure 2.
Features of MCAP syndrome. Photographs of an individual with MCAP syndrome demonstrating the apparent macrocephaly with prominent forehead (D); extensive capillary malformations (A-F1); bilateral 2-3-4 toe syndactyly (G, H); 3-4 finger syndactyly (F1, F2); and postaxial polydactyly of the right hand (F1)
From Mirzaa et al [2012]. Used by permission.

Figure 3.
Features of MCAP syndrome. A boy age 40 months with MCAP syndrome (left) and his unaffected twin sister (right). Note left-sided hemihypertrophy, typical facial features, bilateral 2-3 toe syndactyly, and connective tissue dysplasia with loose redundant skin.
From Conway et al [2007b]. Used by permission.
Overgrowth
PIK3CA pathogenic gain-of-function variants can cause overgrowth of almost any type of tissue. Onset of overgrowth is typically congenital or early postnatal, in contrast to onset in later infancy or childhood.
The predominant areas of involvement include the brain, limbs (including fingers and toes), trunk (including abdomen and chest), and face, all usually in an asymmetric distribution.
Overgrown tissue may have a "ballooning" appearance ‒ that is, the involved body part (usually finger/s, toe/s, and/or dorsum of the hand or foot) resembles an inflated balloon.
Unilateral involvement is more common than bilateral involvement.
Overgrowth may include some or all of the following tissue types:
- Fibrous, including dense fibrous tissue encircling the nerves in individuals with the fibroadipose vascular anomaly (See Table 2.)
- Nervous (See Brain Growth.)
- Vascular (See Vascular Malformations.)
- Lymphatic (See Lymphatic Malformations.)
- Skeletal (See Skeletal Findings.)
Brain Growth
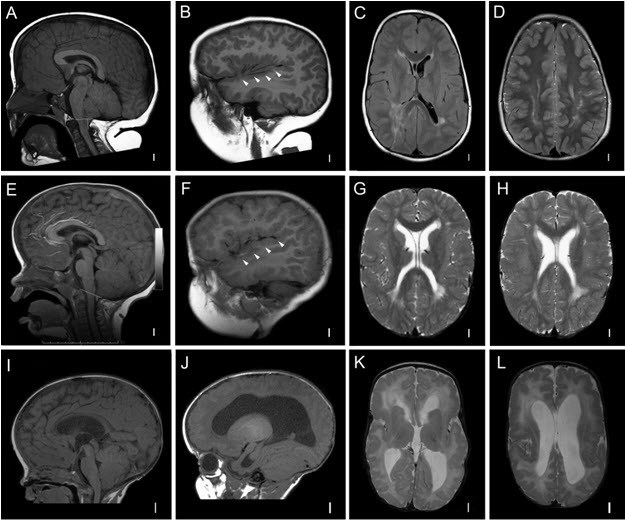
Generalized brain overgrowth may be accompanied by secondary overgrowth of specific brain structures resulting in ventriculomegaly, a markedly thick corpus callosum, and cerebellar tonsillar ectopia with crowding of the posterior fossa [Conway et al 2007b, Mirzaa et al 2012]. Adult OFCs range from +2 to as large as +10 SDs above the mean.
Megalencephalopathy. Although most affected individuals have macrocephaly due to megalencephaly (MEG) at birth, a few affected individuals have normal head size at birth but develop progressive macrocephaly due to progressive MEG within the first year of life [Moore et al 1997, Conway et al 2007a, Conway et al 2007b, Mirzaa et al 2012].
In children who have undergone neurosurgical shunting for obstructive ventriculomegaly or hydrocephalus, head growth noticeably continues at an accelerated pace, indicating the primary nature of MEG in individuals who have MEG as part of their PROS findings.
MCAP syndrome. In a review of 21 children, birth occipitofrontal circumference (OFC) typically ranged from +2 to +7 SDs above the mean for gestational age [Mirzaa et al 2012; Author, unpublished data].
In most children, OFC SD increased during the first year of life. Although head growth may level off in early childhood, it typically remains at +3 SD or more above the mean.
Vascular Malformations
Vascular malformations may include capillary, venous, and less frequently, arterial or mixed (capillary-lymphatic-venous or arteriovenous) malformations. Low-flow vascular malformations (lymphatic, venous) may be found overlying truncal or limb overgrowth. Vascular malformations may be superficial or deep (visceral). Many of these lesions can only be identified by MRA/MRV imaging (see ISSVA Classification for Vascular Anomalies - 2018).
- Cutaneous capillary malformations may be midline on the face (persistent nevus simplex) or widespread on the entire body having a reticulated appearance similar to cutis marmorata.
- Venous malformations (VM), including aneurysms, are characterized by enlarged and distorted blood vessel channels, which may grow over time and cause significant morbidity, including bleeding, pain and disfigurement.
- Affected individuals may be at increased risk for deep venous thrombosis and pulmonary embolism, especially those with combined capillary-lymphatic-venous malformations [Douzgou et al 2022].
- The risk increases after surgery or sclerotherapy.
- Thrombotic risk may also be increased due to other causes of chronic stasis including impaired mobility (e.g., dehydration, surgery), decreased anticoagulant proteins, and the effect of the specific pathogenic PIK3CA variant on the vascular endothelium [Keppler-Noreuil et al 2019].
- High-flow vascular malformations (arteriovenous) can also occur, especially involving the spinal-paraspinal areas.
- Individuals with the Klippel-Trenaunay phenotype can have hemangiomas and venous and/or lymphatic malformations and are at risk for Kasabach-Merritt syndrome, characterized by thrombocytopenia and coagulopathy.
Lymphatic Malformations
Lymphatic malformations may be in various locations (internal and/or external) and can cause various clinical issues including swelling, pain, and occasionally localized bleeding secondary to trauma. Some individuals may have complicated lymphatic anomalies, especially generalized lymphatic anomaly [Rodriguez-Laguna et al 2019]. Complex lymphatic anomalies can have an aggressive course, are difficulty to treat, and have a poor prognosis, especially when located in close proximity to vital structures of the anterior head and neck region.
Skeletal Findings
Characteristic findings in the hands include broad, spade-like hands with splayed or ulnar deviation of the fingers and overgrowth of one or more fingers.
Characteristic findings in the feet include overgrowth with a large "sandal" gap between the great and second toes, large bulbous toes, lipomatous masses on both the dorsal and plantar surfaces, or broad forefoot with wide gaps between the metatarsal heads.
Patterning defects may include postaxial, preaxial, or central polydactyly and cutaneous syndactyly, which often involves the toes, but can include the fingers. The cutaneous syndactyly occurs in patterns of 2-3 toes, 2-4 toes, and 2-5 toes with sandal-gap toes.
Dislocated knees, leg-length discrepancy, and pattern chondromalacia can occur.
Some affected individuals may have scoliosis, vertebral anomalies, spina bifida, and/or pectus anomalies, particularly in the CLOVES phenotype.
Progressive skeletal overgrowth has been described in those with the FH or FAO phenotype.
Individuals with the MCAP phenotype may have joint hypermobility due to connective tissue dysplasia.
Lipomatous Overgrowth with or without Regional Reduction of Adipose Tissue
Lipomatous overgrowth may occur ipsilateral or contralateral to a vascular malformation, if present. The characteristic truncal lipomatous mass infiltrates surrounding tissues and often requires surgical excision. Severe scoliosis, large truncal mass, paraspinal high-flow lesions with spinal cord ischemia, lymphatic malformations, cutaneous vesicles, orthopedic problems of the feet and hands, and central phlebectasia/thromboembolism are examples of significant morbidities that need active or prophylactic medical intervention (see Management).
- Paraspinal and intraspinal extension, more commonly seen in individuals with the CLOVES phenotype, present significant risk for compression of the cord, thecal sac, and nerve roots, with resultant major neurologic deficits including myelopathy, warranting prompt diagnosis and multidisciplinary care [Alomari 2009].
- Lipomatosis can be invasive, invading hip joints and intravertebral spaces, which can become quite painful.
- Infiltration of adipose tissue into muscle with either replacement or compression of muscle, as well as into viscera (liver, spleen, pancreas), intestines, mediastinum, and spine has also been described.
- Surgery can be difficult because of the vascularity of the lipomatous tissue and the risk of thrombosis.
- PIK3CA pathogenic gain-of-function variants can cause regional reduction of adipose tissue that is often accompanied by significant overgrowth in another part of the body. For example, reduction of adipose tissue in the upper limbs, chest, or upper abdomen has been observed in those with significant overgrowth in the lower body or lower limbs/feet.
Developmental Delay and Intellectual Disability
The degree of intellectual disability (ID) appears to be mostly related to the presence and severity of seizures, cortical dysplasia (e.g., polymicrogyria), and hydrocephalus (see Neuroimaging). Gross motor delays are probably attributable to multiple factors in the affected individual including the presence of MEG, cortical brain malformations, hypotonia, limb asymmetry or overgrowth, and connective tissue dysplasia.
MCAP syndrome
- Most individuals with MCAP syndrome have some intellectual disability; the degree is variable and ranges from mild learning disability to severe disability.
- Most have mild-moderate delays yet continue to make steady developmental progress, albeit at a slower rate.
- A few (<10%) have severe handicaps. The range of expected milestone acquisition has not yet been clarified in individuals with MCAP syndrome.
Other Neurodevelopmental Features
Hypotonia. Younger children with polymicrogyria (specifically of the frontal region) have hypotonia, are not spastic, and may have pseudobulbar problems; older children often have spasticity and pseudobulbar problems [Mirzaa et al 2012].
Infant feeding difficulties. Many children may have feeding difficulties that are often multifactorial in nature (e.g., due to hypotonia, GERD), although use of a feeding tube is rarely required.
Epilepsy. An estimated 30%-40% of individuals with PIK3CA pathogenic variants have epilepsy. Reported seizure types include focal and tonic-clonic (among others), though seizure types, severity, age of onset, and any associated EEG or neuroimaging abnormalities depend primarily on the tissue distribution of the pathogenic PIK3CA variants and the presence or absence of associated cortical malformations.
Symptoms of cerebellar tonsillar ectopia (Chiari malformation). Infants may have irritability, excessive drooling, difficulty swallowing, or breathing problems, especially central apnea. Children may have neck pain or headache, motor weakness, sensory changes, vision problems, swallowing difficulties, or behavioral changes.
Behavioral Issues and Autistic Features
A subset of children (6/21) with MCAP syndrome have autistic features or a clinical diagnosis of autism [Mirzaa et al 2012], suggesting that autism may be part of the neurocognitive profile of a minority of individuals who have MEG as part of PROS [McBride et al 2010]. Other behavioral abnormalities seen in one or a few affected individuals:
- Attention-deficit/hyperactivity disorder
- Obsessive-compulsive tendencies
- Anxiety-related issues
Neuroimaging
Individuals with PROS who have macrocephaly typically undergo brain imaging shortly after birth or within the first year of life, leading to early identification of the following key neuroimaging features (see Figure 4) [Vogels et al 1998, Nyberg et al 2005, Conway et al 2007a, Conway et al 2007b, Martínez-Lage et al 2010, Mirzaa et al 2012].
Megalencephaly [Clayton-Smith et al 1997, Vogels et al 1998, Robertson et al 2000, Nyberg et al 2005, Coste et al 2012]. In some instances, MEG (with or without ventriculomegaly) is detected prenatally on ultrasound examination, along with a thickened corpus callosum. More than 90% of affected individuals have congenital MEG that is universally progressive.
Ventriculomegaly and hydrocephalus. Most affected children have evidence of ventricular dilatation or ventriculomegaly on early brain imaging:
- In a large review of the neuroimaging findings in individuals with MCAP syndrome, 37 (56%) of 65 children had ventriculomegaly ranging from mild-to-frank hydrocephalus, with or without cerebellar tonsillar ectopia [Conway et al 2007b].
- While it is unclear whether ventriculomegaly is obstructive in all these individuals, more than half of affected children undergo ventricular shunting or third ventriculostomy, usually within the first year of life.
Cerebellar tonsillar ectopia (CBTE). A large cerebellum combined with a small posterior fossa leading to cerebellar tonsillar ectopia (Chiari malformation) and syringomyelia are common complications, particularly in those with MCAP syndrome:
- Fifteen of 65 reported affected individuals with MCAP have evidence of CBTE with or without herniation [Conway et al 2007b].
- The degree of ectopia is best objectively assessed by measuring the distance of the cerebellar tonsils below the foramen magnum.
- Unlike ventriculomegaly, CBTE is rarely congenital in individuals with MCAP syndrome.
- In two individuals spontaneous "resolution" of CBTE on follow-up imaging was attributed to disproportionately accelerated skull overgrowth [Mirzaa et al 2012].
Cortical brain malformation and polymicrogyria (PMG). PMG may be present in more than 50% of affected children, most commonly those with the MCAP phenotype [Conway et al 2007b; Gripp et al 2009; Mirzaa et al 2012; Authors, unpublished data].
- The most common type of PMG in those with MCAP syndrome is bilateral perisylvian PMG, although other types including bilateral frontal and focal PMG occur.
- PMG broadly, and bilateral perisylvian PMG in particular, increase the risk for: epilepsy; oral motor weakness leading to feeding, swallowing, and expressive language difficulties; developmental delay; and tone abnormalities.
Tumors
Benign tumors. The most common are vascular, described variably as (cavernous) hemangiomas, angiomata, angiomyolipomas, and vascular masses [Clayton-Smith et al 1997, Moore et al 1997, Martínez-Glez et al 2010].
- Cavernous hemangiomas have occurred in brain tissue, necessitating debulking when they enlarge and cause pain.
- While most common in skin or subcutaneous tissue, hemangiomas have been described in viscera and skull.
Other benign tumors have included two individuals with MCAP (ages 21 months and 5 years) who had meningioma (which do not tend to enlarge, spread, or metastasize); two individuals with PROS who had spinal and major nerve neurofibromas; and several others with ovarian cystadenoma, uterine fibroids, and lipomas [Keppler-Noreuil et al 2014].
Malignant tumors. If present, most affected individuals have benign tumors, with only a few malignant tumors reported [Kurek et al 2012, Keppler-Noreuil et al 2014, Luks et al 2015, Gripp et al 2016, Hucthagowder et al 2017, Kuentz et al 2017, Peterman et al 2017, Postema et al 2017].
The estimated frequency of Wilms tumor ranges from 1.4% to 3.3%. Of 12 individuals reported to have Wilms tumor or nephroblastomatosis, clinical PROS diagnoses included CLOVES (8 individuals), MCAP (2 individuals), and KTS (2 individuals).
- Mean age at diagnosis was 27.4 months (median 18 months; range 9-119 months).
- Tumor type included seven individuals (~60%) with Wilms tumor, four (33%) with indeterminate features of Wilms tumor vs nephroblastomatosis, and one (8%) with nephroblastomatosis.
- Six (50%) had somatic "hot spot" PIK3CA variants (see Molecular Genetics).
There have been several case reports of individuals with PROS who developed other cancers including the following [Moore et al 1997, Schwartz et al 2002, Mills et al 2018]:
- Leukemia (in those with the MCAP phenotype)
- Vestibular schwannoma
- Retinoblastoma
Systematic data are at present insufficient to determine whether there is a true association between PROS and the development of these types of tumors or whether the case reports represent rare co-occurrences of PROS with these tumors.
Other
Kidney. Kidney malformations are frequently found in individuals with PROS, and more specifically the CLOVES phenotype, and include pelviectasis, dilated ureters, hydronephrosis, duplicated renal arteries, renal cysts, and enlarged kidneys.
Skin. Abnormalities observed in PROS:
- Dermal melanocytic nevi
- Café au lait macules
- Hypopigmented macules
- Cutis marmorata
- Pigmented nevi
- Patchy hyperpigmentation that follows the lines of Blaschko
- Linear keratinocytic epidermal nevi, which may occur anywhere on the body and may follow a dermatomal distribution
- Seborrheic keratosis and benign lichenoid dermatosis
- Skin hyperelasticity, laxity, and thick subcutaneous tissue in those with the MCAP phenotype due to connective tissue dysplasia
Endocrine issues affect a small number of individuals and most commonly include hypoglycemia (largely hypoinsulinemic hypoketotic hypoglycemia), growth hormone deficiency, and hypothyroidism [Mirzaa et al 2016, Leiter et al 2017, Davis et al 2020, Maines et al 2021, Douzgou et al 2022].
- PROS is a clinical phenocopy of congenital hyperinsulinism (see Molecular Genetics), but plasma insulin concentrations at the time of hypoglycemia are undetectable.
- This profile has been reported both in individuals with MCAP and in those with overlapping forms of PROS that include brain involvement.
- While hypoglycemia in PROS is most commonly diagnosed in the neonatal period, some individuals may present later in childhood, including at least one male with MCAP who presented with his first episode of hypoglycemia at age six years [Author, personal communication].
- Hypoglycemia may be persistent over time, requiring consistent glucose monitoring, evaluation of the hypothalamic-pituitary-adrenal axis, and ongoing treatment (see Management).
Cardiac issues. Structural heart defects (e.g., atrial and ventricular septal defects) and/or abnormalities of the great vessels have been reported in some individuals with MCAP syndrome [Mirzaa et al 2016].
Genotype-Phenotype Correlations
There are several mutational hot spots in PIK3CA (see Molecular Genetics and Table 9) that are more commonly associated with highly focal phenotypes (CLOVES syndrome, fibroadipose hyperplasia, lymphatic/vascular malformations, hemimegalencephaly and focal cortical dysplasia), namely: p.Glu542Lys, p.Glu545Lys, p.His1047Arg, p.His1047Lys [Mirzaa et al 2016]. These mutational hot spots, when present in the brain, are associated with more severe epilepsy phenotypes as well [Pirozzi et al, in press]. The same PIK3CA missense variants may be found in individuals with PROS and in unaffected individuals with PIK3CA-related cancer (Tables 8 and 9).
Further, analysis of individuals with PROS suggests that MCAP syndrome in particular is primarily associated with a wide range of PIK3CA variants that are widely distributed across the gene, and less likely associated with PIK3CA mutational hot spots [Mirzaa et al 2016] (see Molecular Pathogenesis).
Nomenclature
Due to the phenotypic variability of the disorders caused by somatic PIK3CA pathogenic variants, researchers at an NIH Workshop in 2013 proposed the umbrella term PIK3CA-related overgrowth spectrum [Keppler-Noreuil et al 2015]. The PIK3CA-related overgrowth spectrum encompasses all the unique, clinically defined entities but highlights the continuum and overlap between the phenotype diagnoses.
Prevalence
The prevalence of PIK3CA-related overgrowth spectrum (PROS) is difficult to estimate due to variation in ascertainment and the broad phenotypic spectrum. More than 200 individuals with MCAP syndrome have been reported.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in PIK3CA.
Sporadic tumors (including breast, colon, uterine, and others) occurring as single tumors in the absence of any other findings of PROS frequently contain a somatic variant in PIK3CA that is not present in the germline. For more information, see Cancer and Benign Tumors.
Differential Diagnosis
A number of overgrowth and megalencephaly disorders overlap with the PIK3CA-related overgrowth spectrum (PROS), including those summarized in Table 4.
Table 4.
Genes of Interest in the Differential Diagnosis of PIK3CA-Related Overgrowth Spectrum (PROS)
Management
Clinical practice guidelines for PIK3CA-related overgrowth spectrum have been published [Douzgou et al 2022] (full text). Additionally, a targeted pharmacologic therapy has been FDA approved (see Table 6).
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual with PIK3CA-related overgrowth spectrum (PROS), the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Note: Assessment is complicated by variable findings in individuals with this condition. Accurate and thorough assessment of medical history is necessary to evaluate for vascular malformations as well as other clinical features.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with PIK3CA-Related Overgrowth Spectrum
Treatment of Manifestations
Targeted Therapy
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
There is no cure for PIK3CA-related overgrowth syndrome (PROS). Table 6 details a targeted pharmacologic treatment recently approved by the FDA.
Table 6.
Targeted Treatment of PIK3CA-Related Overgrowth Spectrum
Supportive Care
Supportive treatment should ideally by provided through coordinated care from a multidisciplinary team including surgeons, radiologists, geneticists, dermatologists, pathologists, and hematologist/oncologist, the latter of whom are critical for emerging medical management and coordination of the associated long-term follow up [Adams & Ricci 2019, Dekeuleneer et al 2020, Canaud et al 2021, Douzgou et al 2022].
Table 7.
Supportive Treatment of Manifestations in Individuals with PIK3CA-Related Overgrowth Spectrum
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, Botox®, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one-on-one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Table 8.
Recommended Surveillance for Individuals with PIK3CA-Related Overgrowth Spectrum
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Some studies have demonstrated the efficacy of the mammalian target of rapamycin (mTOR) inhibitor sirolimus for lymphatic diseases [Padilla et al 2019, Zenner et al 2019]. Ongoing understanding of the PI3K/AKT/mTOR signaling pathway and the natural history of PROS will provide the basis for developing new targeted therapeutic strategies. Systemic agents under investigation for PROS target different components of the PI3K signaling pathway. These include inhibitors of mTOR, AKT, and PI3K genes:
- Sirolimus has been investigated in several clinical trials in individuals with complex lymphatic anomalies, vascular malformations, and overgrowth disorders [Adams et al 2016, Hammer et al 2018, Adams & Ricci 2019, Parker et al 2019, Van Damme et al 2020] (see ClinicalTrials.gov), some of whom had PROS. Sirolimus is currently used in an off-label capacity to treat these disorders [Seront et al 2019, Dekeuleneer et al 2020]. Sirolimus has shown efficacy in multiple Phase II studies for individuals with complicated vascular anomalies and individuals with PROS and progressive overgrowth [Adams et al 2016, Erickson et al 2017, Hammer et al 2018, Parker et al 2019, Ricci et al 2019].
- Miransertib, an AKT1 inhibitor, has been studied in clinical trials for expanded access, and in Phase I/II clinical trials [Wassef et al 2015, Zampino et al 2019].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
PIK3CA-related overgrowth disorders (PROS) are not known to be inherited, as most identified pathogenic variants are somatic (mosaic). No confirmed vertical transmission or sib recurrence has been reported to date.
Risk to Family Members
Parents of a proband
- Parents of children with somatic mosaicism for a pathogenic variant in PIK3CA have not been reported to have any significant distinctive manifestations of the disorder, nor would such findings be expected given the somatic nature of these genetic alterations.
- Theoretically, a parent of a child with PROS caused by a de novo germline PIK3CA pathogenic variant may have germline mosaicism for the PIK3CA pathogenic variant. The theoretic risk for parental germline mosaicism is estimated to be less than 1%.
Sibs of a proband
- The risk to sibs of a proband with somatic mosaicism for a pathogenic variant in PIK3CA would be expected to be the same as in the general population.
- The risk to sibs of a proband with a de novo germline PIK3CA pathogenic variant is slightly greater than that of the general population because of the theoretic risk (estimated to be <1%) of parental germline mosaicism.
Offspring of a proband
- Reproductive outcome data on adults with PROS are limited; there are no instances of vertical transmission of these disorders. While adults with PROS have been reported, the developmental outcome of affected individuals is unknown. Individuals with significant neurologic involvement (e.g., DMEG, HMEG) have a poor prognosis.
- All but a few affected individuals with PROS have had somatic mosaicism for a PIK3CA pathogenic variant, suggesting that mutation occurred post fertilization in one cell of the multicellular embryo. Therefore, the risk for transmission to offspring is expected to be less than 50%.
- Several individuals with PROS have had a de novo germline pathogenic variant in PIK3CA [Rivière et al 2012, Mirzaa et al 2016]. The offspring of individuals with a constitutional germline PIK3CA pathogenic variant have a 50% risk of inheriting the pathogenic variant.
Other family members. The risk to other family members is the same as that of the general population.
Related Genetic Counseling Issues
Family planning
- Counseling for recurrence risk in PROS should emphasize that, while no pregnancy is at zero risk, all empiric evidence suggests that the risk for recurrence in sibs of a proband is not increased over that of the general population, due to the mosaic nature of most of these disorders. The rare families with PROS caused by a de novo germline PIK3CA pathogenic variant should be counseled regarding the theoretic risk for parental germline mosaicism (~<1%).
- The optimal time for determination of genetic risk is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Because vertical transmission of PROS has not been reported to date, family members are not known to be at increased risk of being affected and prenatal diagnosis and preimplantation genetic testing are usually not indicated for family members. However, low-level germline mosaicism may theoretically be present in a parent of a child with a germline PIK3CA pathogenic variant. Prenatal testing or preimplantation genetic testing may be an option for such rare families.
In addition, molecular genetic prenatal testing may be an option for pregnancies identified by ultrasound examination to be at risk for PROS.
- Findings on ultrasound examination that suggest MCAP syndrome include marked fetal overgrowth and progressive macrocephaly with no indication of maternal hyperglycemia or fetal hyperinsulinism. Other reported fetal ultrasound findings include ventriculomegaly, pleural effusions, polyhydramnios, hydrops, limb asymmetry, and frontal bossing [Nyberg et al 2005]. A fetal MRI in an affected fetus with megalencephaly and ventriculomegaly revealed diffuse bilateral polymicrogyria and polysyndactyly of one foot [Gripp et al 2009].
- Findings on prenatal ultrasound examination in CLOVES syndrome include prenatal overgrowth, lipomatous truncal masses, and vascular or lymphatic malformations [Sapp et al 2007, Alomari 2009, Alomari 2011].
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- CLOVES Syndrome CommunityPO BOX 406West Kennebunk 04094Phone: 833-425-6837Email: info@clovessyndrome.org
- M-CM NetworkPO Box 97Chatham NY 12037Phone: 518-392-2150Email: hello@m-cm.net
- M-CM Network Contact RegistryThe M-CM Network contact registry will be used to inform individuals with M-CM and their guardians about: opportunities to participate in research, opportunities to contribute data, and discoveries about M-CM that may impact care decisions.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
PIK3CA-Related Overgrowth Spectrum: Genes and Databases
Table B.
OMIM Entries for PIK3CA-Related Overgrowth Spectrum (View All in OMIM)
Molecular Pathogenesis
PIK3CA-related overgrowth spectrum disorders are typically caused by postzygotic somatic variants in the gene that encodes phosphatidylinositol-3-kinase (PI3K) catalytic subunit alpha (p110alpha) [Engelman et al 2006]. These variants are associated with hyperactivation of the PI3K signaling pathway, which includes multiple downstream effectors such as AKT and mTOR, resulting in abnormal growth of various tissues and vascular malformations.
The PIK3CA protein is critical for the action of insulin to lower blood glucose, and for the action of insulin-like growth factor 1 (IGF1), which promotes tissue growth through a receptor closely similar to the insulin receptor, and which mediates many actions of growth hormone. Pathologically activated PIK3CA may mimic the action of insulin and/or IGF1 in cells. For such a mechanism to translate into a clinically important endocrinopathy, target tissues need to have a high variant burden. This mechanism may explain why some affected infants have clinical features similar to hyperinsulinism.
Mechanism of disease causation. Gain of function
PIK3CA-specific laboratory technical considerations. Because most affected individuals having a mosaic PIK3CA pathogenic variant, use of custom restriction fragment length polymorphism (RFLP) assays or digital droplet PCR on an appropriate sample may be necessary. Standard-depth exome sequencing can miss mosaic PIK3CA variants, especially if performed on blood-derived DNA.
- In order to detect new or very rare variants, sequencing of entire exons is typically necessary.
- Sanger sequencing can be used only if the pathogenic variant allele fraction is relatively high (~20%).
- Targeted capture of the entire PIK3CA coding region followed by next-generation sequencing at very deep coverage may be better suited for somatic variant detection, as it allows for detection of very low levels of mosaicism throughout the gene.
- In MCAP syndrome, sequence analysis of DNA derived from saliva or skin fibroblasts (whether visibly affected or not) has a higher detection rate than peripheral blood-derived DNA [Mirzaa et al 2016].
- In focal brain overgrowth disorders (HMEG, FCD, DMEG), analysis of DNA derived from affected brain tissues (for example, removed at the time of epilepsy surgery) has a higher detection rate than peripheral tissues (blood or saliva) [Jansen et al 2015].
Table 9.
Notable PIK3CA Pathogenic Variants
Cancer and Benign Tumors
PIK3CA is somatically mutated in many cancers including colorectal, ovarian, breast, hepatocellular carcinomas, and glioblastomas. These PIK3CA pathogenic variants are located mostly at hot spots within the kinase domain (encoded by exon 20), and result in gain of function implicated in oncogenicity [Samuels et al 2004, Ikenoue et al 2005, Kang et al 2005].
Most (>80%) activating PIK3CA pathogenic variants in cancer (Table 10) and PROS (Table 9) cluster at three hot spots: two glutamic acid (Glu) residues at codons 542 and 545, and a histidine (His) residue at codon 1047. The distribution of PIK3CA pathogenic variants in cancer was obtained from the Catalogue of Somatic Mutations in Cancer (COSMIC, v85, May 2018). PIK3CA pathogenic variants in PROS comprise published cases from larger cohort studies. The risk for tumorigenesis and development of malignancies is a theoretic concern because PIK3CA is somatically mutated or overexpressed in many cancers. The pathogenic variants in PIK3CA in cancers and in a large proportion of individuals with PROS (especially those with the phenotypes of CLOVES, fibroadipose hyperplasia, and isolated macrodactyly) are most commonly located at hot spots within the helical and kinase domains.
Among the approximately 160 PIK3CA pathogenic variants listed in the Catalogue of Somatic Mutations in Cancer (COSMIC), common variants in three amino acids (p.Glu542Lys, p.Glu545Lys in exon 9, and p.His1047Arg and p.His1047Leu in exon 20) account for 80% of tumor-associated PIK3CA variants and show the highest oncogenic activity [Samuels et al 2004, Samuels & Ericson 2006, Janku et al 2012]. See Table 10.
Table 10.
Common Cancer-Related PIK3CA Variants Reported in COSMIC Database
Chapter Notes
Author Notes
Dr Ghayda Mirzaa is an Associate Professor of Medical Genetics and Pediatrics at the University of Washington School of Medicine. Her research is focused on developmental brain disorders including megalencephaly with multiple publications related to PIK3CA-related overgrowth spectrum (PROS) including the natural history, molecular diagnosis, and potential therapies.
Dr John M Graham Jr is a Professor Emeritus of Pediatrics at David Geffen School of Medicine at UCLA and Consulting Clinical Geneticist at Cedars-Sinai Medical Center and Harbor UCLA Medical Center. He has had a long-term clinical interest in PROS with many publications on this topic.
Dr Kim Keppler-Noreuil is a clinical geneticist and Division Chief of Genetics & Metabolism at the University of Wisconsin School of Medicine and Public Health; she has multiple publications related to PROS, including invited review articles and original research focused particularly on the clinical and molecular diagnosis, and potential therapies for PROS.
Acknowledgments
We would like to thank the patients, their families, and our collaborators for their valuable contribution to our knowledge about these disorders.
Author History
Robert Conway, MD; Wayne State University (2013-2021)
Willliam B Dobyns, MD; Seattle Childfren's Hospital (2013-2021)
John H Graham Jr, MD, ScD (2013-present)
Kim Keppler-Noreuil, MD, FAAP, FACMG (2021-present)
Ghayda Mirzaa, MD, FAAP, FACMG (2013-present)
Revision History
- 6 April 2023 (ma/aa) Revision: expanded information on management of hypoglycemia; Targeted Therapy linked as a Key Section
- 25 August 2022 (aa) Revision: FDA approval of alpelisib for treatment of PROS (Table 6)
- 23 December 2021 (ma) Comprehensive update posted live
- 15 August 2013 (me) Review posted live
- 11 March 2013 (gm) Original submission
References
Literature Cited
- Adams DM, Ricci KW. Vascular anomalies: diagnosis of complicated anomalies and new medical treatment options. Hematol Oncol Clin North Am. 2019;33:455-70. [PubMed: 31030813]
- Adams DM, Trenor CC, 3rd, Hammill AM, Vinks AA, Patel MN, Chaudry G, Wentzel MS, Mobberley-Schuman PS, Campbell LM, Brookbank C, Gupta A, Chute C, Eile J, McKenna J, Merrow AC, Fei L, Hornung L, Seid M, Dasgupta AR, Dickie BH, Elluru RG, Lucky AW, Weiss B, Azizkhan RG. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. 2016;137:e20153257. [PMC free article: PMC4732362] [PubMed: 26783326]
- Alomari AI. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: a descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol. 2009;18:1-7. [PubMed: 19011570]
- Alomari AI. Comments on the diagnosis and management of CLOVES syndrome. Pediatr Dermatol. 2011;28:215-6. [PubMed: 21504461]
- Canaud G, Hammill AM, Adams D, Vikkula M, Keppler-Noreuil KM. 2021. A review of mechanisms of disease across PIK3CA-related disorder with vascular malformations. Orphanet J Rare Dis 16:306. [PMC free article: PMC8268514] [PubMed: 34238334]
- Clayton-Smith J, Kerr B, Brunner H, Tranebjaerg L, Magee A, Hennekam RC, Mueller RF, Brueton L, Super M, Steen-Johnsen J, Donnai D. Macrocephaly with cutis marmorata, haemangioma and syndactyly--a distinctive overgrowth syndrome. Clin Dysmorphol. 1997;6:291-302. [PubMed: 9354837]
- Conway RL, Danielpour M, Graham JM Jr. Surgical management of cerebellar tonsillar herniation in three patients with macrocephaly-cutis marmorata telangiectatica congenita. Report of three cases. J Neurosurg. 2007a;106:296-301. [PubMed: 17465364]
- Conway RL, Pressman BD, Dobyns WB, Danielpour M, Lee J, Sanchez-Lara PA, Butler MG, Zackai E, Campbell L, Saitta SC, Clericuzio CL, Milunsky JM, Hoyme HE, Shieh J, Moeschler JB, Crandall B, Lauzon JL, Viskochil DH, Harding B, Graham JM Jr. Neuroimaging findings in macrocephaly-capillary malformation: a longitudinal study of 17 patients. Am J Med Genet A. 2007b;143A:2981-3008. [PMC free article: PMC6816457] [PubMed: 18000912]
- Coste K, Sarret C, Cisse A, Delabaere A, Francannet C, Vanlieferinghen P. Macrocephaly-capillary malformation. A neonatal case Arch Pediatr. 2012;19:917-20. [PubMed: 22884750]
- Couto JA, Konczyk DJ, Vivero MP, Kozakewich HPW, Upton J, Fu X, Padwa BL, Mulliken JB, Warman ML, Greene AK. Somatic PIK3CA mutations are present in multiple tissues of facial infiltrating lipomatosis. Pediatr Res. 2017;82:850-4. [PMC free article: PMC5645230] [PubMed: 28665924]
- Davis S, Ware MA, Zeiger J, Deardorff MA, Grand K, Grimberg A, Hsu S, Kelsey M, Majidi S, Matthew RP, Napier M, Nokoff N, Prasad C, Riggs AC, McKinnon ML, Mirzaa G. Growth hormone deficiency in megalencephaly-capillary malformation syndrome: An association with activating mutations in PIK3CA. Am J Med Genet A. 2020;182:162-8. [PMC free article: PMC7262792] [PubMed: 31729162]
- Dekeuleneer V, Seront E, Van Damme A, Boon LM, Vikkula M. Theranostic advances in vascular malformations. J Invest Dermatol. 2020;140:756-63. [PubMed: 32200879]
- D'Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, Hossain A, Hatem NE, Barry BJ, Kwiatkowski DJ, Vinters HV, Barkovich AJ, Shendure J, Mathern GW, Walsh CA, Poduri A. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77:720-5. [PMC free article: PMC4471336] [PubMed: 25599672]
- Di Rocco C, Battaglia D, Pietrini D, Piastra M, Massimi L. Hemimegalencephaly: clinical implications and surgical treatment. Childs Nerv Syst. 2006;22:852-66. [PubMed: 16821075]
- Douzgou S, Rawson M, Baselga E, Danielpour M, Faivre L, Kashanian A, Keppler-Noreuil KM, Kuentz P, Mancini GMS, Maniere MC, Martinez-Glez V, Parker VE, Semple RK, Srivastava S, Vabres P, De Wit MY, Graham JM Jr, Clayton-Smith J, Mirzaa GM, Biesecker LG. A standard of care for individuals with PIK3CA-related disorders: an international expert consensus statement. Clin Genet. 2022;101:32-47. [PMC free article: PMC8664971] [PubMed: 34240408]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606-19. [PubMed: 16847462]
- Erickson J, McAuliffe W, Blennerhassett L, Halbert A. Fibroadipose vascular anomaly treated with sirolimus: successful outcome in two patients. Pediatr Dermatol. 2017;34:e317-e320 [PubMed: 29144050]
- Gripp KW, Baker L, Kandula V, Conard K, Scavina M, Napoli JA, Griffin GC, Thacker M, Knox RG, Clark GR, Parker VE, Semple R, Mirzaa G, Keppler-Noreuil KM. Nephroblastomatosis or Wilms tumor in a fourth patient with a somatic PIK3CA mutation. Am J Med Genet A. 2016;170:2559-69. [PMC free article: PMC5514817] [PubMed: 27191687]
- Gripp KW, Hopkins E, Vinkler C, Lev D, Malinger G, Lerman-Sagie T, Dobyns WB. Significant overlap and possible identity of macrocephaly capillary malformation and megalencephaly polymicrogyria-polydactyly hydrocephalus syndromes. Am J Med Genet A. 2009;149A:868-76. [PubMed: 19353582]
- Hammer J, Seront E, Duez S, Dupont S, Van Damme A, Schmitz S, Hoyoux C, Chopinet C, Clapuyt P, Hammer F, Vikkula M, Boon LM. Sirolimus is efficacious in treatment for extensive and/or complex slow-flow vascular malformations: a monocentric prospective phase II study. Orphanet J Rare Dis. 2018;13:191. [PMC free article: PMC6206885] [PubMed: 30373605]
- Hucthagowder V, Shenoy A, Corliss M, Vigh-Conrad KA, Storer C, Grange DK, Cottrell CE. Utility of clinical high-depth next generation sequencing for somatic variant detection in the PIK3CA-related overgrowth spectrum. Clin Genet. 2017;91:79-85. [PubMed: 27307077]
- Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, Kawabe T, Omata M. 2005. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 65:4562–7. [PubMed: 15930273]
- Janku F, Wheler JJ, Naing A, Stepanek VM, Falchook GS, Fu S, Garrido-Laguna I, Tsimberidou AM, Piha-Paul SA, Moulder SL, Lee JJ, Luthra R, Hong DS, Kurzrock R. PIK3CA mutations in advanced cancers: characteristics and outcomes. Oncotarget. 2012;3:1566-75. [PMC free article: PMC3681495] [PubMed: 23248156]
- Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ, Hiatt JB, Roden WH, Gunter SA, Christian SL, Collins S, Adams C, Rivière JB, St-Onge J, Ojemann JG, Shendure J, Hevner RF, Dobyns WB. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. 2015;138:1613-28. [PMC free article: PMC4614119] [PubMed: 25722288]
- Kang S, Bader AG, Vogt PK. 2005. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA 102:802–7. [PMC free article: PMC545580] [PubMed: 15647370]
- Keppler-Noreuil KM, Lozier J, Oden N, Taneja A, Burton-Akright J, Sapp JC, Biesecker LG. Thrombosis risk factors in PIK3CA-related overgrowth spectrum and Proteus syndrome. Am J Med Genet C Semin Med Genet. 2019;181:571-81. [PMC free article: PMC8513083] [PubMed: 31490637]
- Keppler-Noreuil KM, Rios JJ, Parker VE, Semple RK, Lindhurst MJ, Sapp JC, Alomari A, Ezaki M, Dobyns W, Biesecker LG. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. 2015;167A:287-95. [PMC free article: PMC4480633] [PubMed: 25557259]
- Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Parker VE, Blumhorst C, Darling T, Tosi LL, Huson SM, Whitehouse RW, Jakkula E, Grant I, Balasubramanian M, Chandler KE, Fraser JL, Gucev Z, Crow YJ, Brennan LM, Clark R, Sellars EA, Pena LD, Krishnamurty V, Shuen A, Braverman N, Cunningham ML, Sutton VR, Tasic V, Graham JM Jr, Geer J Jr, Henderson A, Semple RK, Biesecker LG. Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. Am J Med Genet A. 2014;164A:1713-33. [PMC free article: PMC4320693] [PubMed: 24782230]
- Kuentz P, St-Onge J, Duffourd Y, Courcet JB, Carmignac V, Jouan T, Sorlin A, Abasq-Thomas C, Albuisson J, Amiel J, Amram D, Arpin S, Attie-Bitach T, Bahi-Buisson N, Barbarot S, Baujat G, Bessis D, Boccara O, Bonnière M, Boute O, Bursztejn AC, Chiaverini C, Cormier-Daire V, Coubes C, Delobel B, Edery P, Chehadeh SE, Francannet C, Geneviève D, Goldenberg A, Haye D, Isidor B, Jacquemont ML, Khau Van Kien P, Lacombe D, Martin L, Martinovic J, Maruani A, Mathieu-Dramard M, Mazereeuw-Hautier J, Michot C, Mignot C, Miquel J, Morice-Picard F, Petit F, Phan A, Rossi M, Touraine R, Verloes A, Vincent M, Vincent-Delorme C, Whalen S, Willems M, Marle N, Lehalle D, Thevenon J, Thauvin-Robinet C, Hadj-Rabia S, Faivre L, Vabres P, Rivière JB. Molecular diagnosis of PIK3CA-related overgrowth spectrum (PROS) in 162 patients and recommendations for genetic testing. Genet Med. 2017;19:989-97. [PubMed: 28151489]
- Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, Mulliken JB, Bowen ME, Yamamoto GL, Kozakewich HP, Warman ML. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108-15. [PMC free article: PMC3370283] [PubMed: 22658544]
- Kwan SY, Shyu HY, Lin JH, Wong TT, Chang KP, Yiu CH. Corpus callosotomy in a patient of hemimegalencephaly and Lennox-Gastaut syndrome. Brain Dev. 2008;30:643-6. [PubMed: 18439776]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941-5. [PMC free article: PMC4417942] [PubMed: 22729223]
- Leiter SM, Parker VER, Welters A, Knox R, Rocha N, Clark G, Payne F, Lotta L, Harris J, Guerrero-Fernández J, González-Casado I, García-Miñaur S, Gordo G, Wareham N, Martínez-Glez V, Allison M, O'Rahilly S, Barroso I, Meissner T, Davies S, Hussain K, Temple K, Barreda-Bonis AC, Kummer S, Semple RK. Hypoinsulinaemic, hypoketotic hypoglycaemia due to mosaic genetic activation of PI3-kinase. Eur J Endocrinol. 2017;177:175-86. [PMC free article: PMC5488397] [PubMed: 28566443]
- Lindhurst MJ, Parker VE, Payne F, Sapp JC, Rudge S, Harris J, Witkowski AM, Zhang Q, Groeneveld MP, Scott CE, Daly A, Huson SM, Tosi LL, Cunningham ML, Darling TN, Geer J, Gucev Z, Sutton VR, Tziotzios C, Dixon AK, Helliwell T, O'Rahilly S, Savage DB, Wakelam MJ, Barroso I, Biesecker LG, Semple RK. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928-33. [PMC free article: PMC3461408] [PubMed: 22729222]
- Luks VL, Kamitaki N, Vivero MP, Uller W, Rab R, Bovée JV, Rialon KL, Guevara CJ, Alomari AI, Greene AK, Fishman SJ, Kozakewich HP, Maclellan RA, Mulliken JB, Rahbar R, Spencer SA, Trenor CC 3rd, Upton J, Zurakowski D, Perkins JA, Kirsh A, Bennett JT, Dobyns WB, Kurek KC, Warman ML, McCarroll SA, Murillo R. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166:1048-54.e1. [PMC free article: PMC4498659] [PubMed: 25681199]
- Maines E, Franceschi R, Martinelli D, Soli F, Lepri FR, Piccoli G, Soffiati M. Hypoglycemia due to PI3K/AKT/mTOR signaling pathway defects: two novel cases and review of the literature. Hormones (Athens). 2021;20:623-40. [PubMed: 33876391]
- Martínez-Glez V, Romanelli V, Mori MA, Gracia R, Segovia M, González-Meneses A, López-Gutierrez JC, Gean E, Martorell L, Lapunzina P. Macrocephaly-capillary malformation: Analysis of 13 patients and review of the diagnostic criteria. Am J Med Genet A. 2010;152A:3101-6. [PubMed: 21077203]
- Martínez-Lage JF, Guillén-Navarro E, Almagro MJ, Felipe-Murcia M, López López-Guerrero A, Galarza M. Hydrocephalus and Chiari type 1 malformation in macrocephaly-cutis marmorata telangiectatica congenita: a case-based update. Childs Nerv Syst. 2010;26:13-8. [PubMed: 19763591]
- McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, Herman GE. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3:137-41. [PubMed: 20533527]
- Mills JR, Moyer AM, Kipp BR, Poplawski AB, Messiaen LM, Babovic-Vuksanovic D. Unilateral vestibular schwannoma and meningiomas in a patient with PIK3CA-related segmental overgrowth: Co-occurrence of mosaicism for 2 rare disorders. Clin Genet. 2018;93:187-90. [PubMed: 28737257]
- Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries LS, Lev D, Kramer N, Hopkins E, Graham JM Jr, Dobyns WB. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A:269-91. [PubMed: 22228622]
- Mirzaa G, Timms AE, Conti V, Boyle EA, Girisha KM, Martin B, Kircher M, Olds C, Juusola J, Collins S, Park K, Carter M, Glass I, Krägeloh-Mann I, Chitayat D, Parikh AS, Bradshaw R, Torti E, Braddock S, Burke L, Ghedia S, Stephan M, Stewart F, Prasad C, Napier M, Saitta S, Straussberg R, Gabbett M, O'Connor BC, Keegan CE, Yin LJ, Lai AHM, Martin N, McKinnon M, Addor MC, Boccuto L, Schwartz CE, Lanoel A, Conway RL, Devriendt K, Tatton-Brown K, Pierpont ME, Painter M, Worgan L, Reggin J, Hennekam R, Tsuchiya K, Pritchard CC, Aracena M, Gripp KW, Cordisco M, Van Esch H, Garavelli L, Curry C, Goriely A, Kayserilli H, Shendure J, Graham J Jr, Guerrini R, Dobyns WB. PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. JCI Insight. 2016;1:e87623. [PMC free article: PMC5019182] [PubMed: 27631024]
- Moore CA, Toriello HV, Abuelo DN, Bull MJ, Curry CJ, Hall BD, Higgins JV, Stevens CA, Twersky S, Weksberg R, Dobyns WB. Macrocephaly-cutis marmorata telangiectatica congenita: a distinct disorder with developmental delay and connective tissue abnormalities. Am J Med Genet. 1997;70:67-73. [PubMed: 9129744]
- Nyberg RH, Uotila J, Kirkinen P, Rosendahl H. Macrocephaly-cutis marmorata telangiectatica congenita syndrome--prenatal signs in ultrasonography. Prenat Diagn. 2005;25:129-32. [PubMed: 15712320]
- Padilla CA, Bárcena JA, López-Grueso MJ, Requejo-Aguilar R. The regulation of TORC1 pathway by the yeast chaperones Hsp31 is mediated by SFP1 and affects proteasomal activity. Biochim Biophys Acta Gen Subj. 2019;1863:534-46. [PubMed: 30578832]
- Parker VE, Keppler-Noreuil KM, Faivre L, Luu M, Oden NL, De Silva L, Sapp JC, Andrews K, Bardou M, Chen KY, Darling TN, Gautier E, Goldspiel BR, Hadj-Rabia S, Harris J, Kounidas G, Kumar P, Lindhurst MJ, Loffroy R, Martin L, Phan A, Rother KI, Widemann BC, Wolters PL, Coubes C, Pinson L, Willems M, Vincent-Delorme C, Vabres P, Semple RK, Biesecker LG, et al. Safety and efficacy of low-dose sirolimus in the PIK3CA-related overgrowth spectrum. Genet Med. 2019;21:1189-98. [PMC free article: PMC6752269] [PubMed: 30270358]
- Peterman CM, Fevurly RD, Alomari AI, Trenor CC 3rd, Adams DM, Vadeboncoeur S, Liang MG, Greene AK, Mulliken JB, Fishman SJ. Sonographic screening for Wilms tumor in children with CLOVES syndrome. Pediatr Blood Cancer. 2017;64. [PubMed: 28627003]
- Pirozzi F, Berkseth M, Shear R, Gonzalez L, Timms AE, Sulc J, Pao E, Oyama N, Forzano F, Conti V, Guerrini R, Doherty ES, Saitta SC, Lockwood CM, Pritchard CC, Dobyns WB, Novotny E, Wright J, Saneto RP, Friedman S, Hauptman J, Ojemann J, Kapur RP, Mirzaa GMM. Profiling PI3K-AKT-MTOR variants in focal brain malformations reveals new insights for diagnostic care. Brain. In press. [PMC free article: PMC9630661] [PubMed: 35355055]
- Postema FAM, Hopman SMJ, Aalfs CM, Berger LPV, Bleeker FE, Dommering CJ, Jongmans MCJ, Letteboer TGW, Olderode-Berends MJW, Wagner A, Hennekam RC, Merks JHM. Childhood tumours with a high probability of being part of a tumour predisposition syndrome; reason for referral for genetic consultation. Eur J Cancer. 2017;80:48-54. [PubMed: 28544908]
- Ricci KW, Hammill AM, Mobberley-Schuman P, Nelson SC, Blatt J, Bender JLG, McCuaig CC, Synakiewicz A, Frieden IJ, Adams DM. Efficacy of systemic sirolimus in the treatment of generalized lymphatic anomaly and Gorham-Stout disease. Pediatr Blood Cancer. 2019;66:e27614. [PMC free article: PMC6428616] [PubMed: 30672136]
- Rivière JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GM, Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, Uyanik G, Weksberg R, Zirn B, Beaulieu CL; Finding of Rare Disease Genes (FORGE) Canada Consortium, Majewski J, Bulman DE, O'Driscoll M, Shendure J, Graham JM Jr, Boycott KM, Dobyns WB. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934-40. [PMC free article: PMC3408813] [PubMed: 22729224]
- Robertson SP, Gattas M, Rogers M, Adès LC. Macrocephaly--cutis marmorata telangiectatica congenita: report of five patients and a review of the literature. Clin Dysmorphol. 2000;9:1-9. [PubMed: 10649789]
- Rodriguez-Laguna L, Agra N, Ibañez K, Oliva-Molina G, Gordo G, Khurana N, Hominick D, Beato M, Colmenero I, Herranz G, Torres Canizalez JM, Rodríguez Pena R, Vallespín E, Martín-Arenas R, Del Pozo Á, Villaverde C, Bustamante A, Ayuso C, Lapunzina P, Lopez-Gutierrez JC, Dellinger MT, Martinez-Glez V. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J Exp Med. 2019;216:407-18. [PMC free article: PMC6363432] [PubMed: 30591517]
- Sadick M, Müller-Wille R, Wildgruber M, Wohlgemuth WA. Vascular anomalies (part i): classification and diagnostics of vascular anomalies. Rofo. 2018;190:825-35. [PubMed: 29874693]
- Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77-82. [PubMed: 16357568]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. [PubMed: 15016963]
- Sapp JC, Turner JT, van de Kamp JM, van Dijk FS, Lowry RB, Biesecker LG. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am J Med Genet A. 2007;143A:2944-58. [PubMed: 17963221]
- Schwartz IVD, Felix TM, Riegel M, Schüler-Faccini L. Atypical macrocephaly-cutis marmorata telangiectatica congenita with retinoblastoma. Clin Dysmorphol. 2002;11:199-202. [PubMed: 12072801]
- Seront E, Van Damme A, Boon LM, Vikkula M. Rapamycin and treatment of venous malformations. Curr Opin Hematol. 2019;26:185-92. [PubMed: 30855337]
- Van Damme A, Seront E, Dekeuleneer V, Boon LM, Vikkula M. New and emerging targeted therapies for vascular malformations. Am J Clin Dermatol. 2020;21:657-68. [PubMed: 32557381]
- Vogels A, Devriendt K, Legius E, Decock P, Marien J, Hendrickx G, Fryns JP. The macrocephaly-cutis marmorata telangiectatica congenita syndrome. Long-term follow-up data in 4 children and adolescents. Genet Couns. 1998;9:245-53. [PubMed: 9894160]
- Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, Burrows P, Frieden IJ, Garzon MC, Lopez-Gutierrez JC, Lord DJ, Mitchel S, Powell J, Prendiville J, Vikkula M, et al. Vascular anomalies classification: recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics. 2015;136:e203-14. [PubMed: 26055853]
- Zampino G, Leoni C, Buonuomo PS, Rana I, Onesimo R, Macchiaiolo M, et al. An open-label, Phase I/II study of miransertib (ARQ 092), an oral pan-AKT inhibitor, in patients (pts) with PIK3CA-related overgrowth spectrum (PROS) and Proteus syndrome (PS): study design and preliminary results (NCT03094832). Gothenburg, Sweden: European Society of Human Genetics Conference; June 15-18, 2019; Abstract C 18.6.
- Zenner K, Cheng CV, Jensen DM, Timms AE, Shivaram G, Bly R, Ganti S, Whitlock KB, Dobyns WB, Perkins J, Bennett JT. Genotype correlates with clinical severity in PIK3CA-associated lymphatic malformations. JCI Insight. 2019;4:e129884. [PMC free article: PMC6948764] [PubMed: 31536475]
Publication Details
Author Information and Affiliations
Seattle, Washington
Los Angeles, California
Madison, Wisconsin
Publication History
Initial Posting: August 15, 2013; Last Revision: April 6, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Mirzaa G, Graham JM Jr, Keppler-Noreuil K. PIK3CA-Related Overgrowth Spectrum. 2013 Aug 15 [Updated 2023 Apr 6]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.