Summary
Clinical characteristics.
Phosphorylase kinase (PhK) deficiency causing glycogen storage disease type IX (GSD IX) results from deficiency of the enzyme phosphorylase b kinase, which has a major regulatory role in the breakdown of glycogen. The two types of PhK deficiency are liver PhK deficiency (characterized by early childhood onset of hepatomegaly and growth restriction, and often, but not always, fasting ketosis and hypoglycemia) and muscle PhK deficiency, which is considerably rarer (characterized by any of the following: exercise intolerance, myalgia, muscle cramps, myoglobinuria, and progressive muscle weakness). While symptoms and biochemical abnormalities of liver PhK deficiency were thought to improve with age, it is becoming evident that affected individuals need to be monitored for long-term complications such as liver fibrosis and cirrhosis.
Diagnosis/testing.
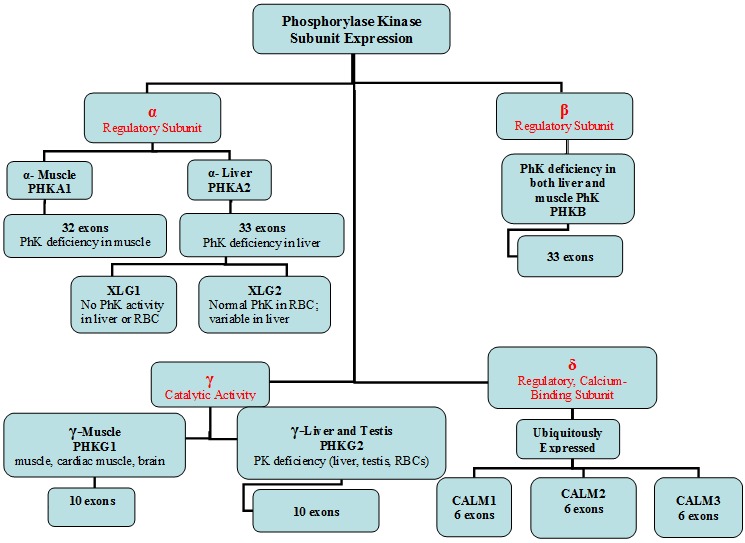
The enzyme PhK comprises four copies each of four subunits (α, β, γ, and δ).
Pathogenic variants in:
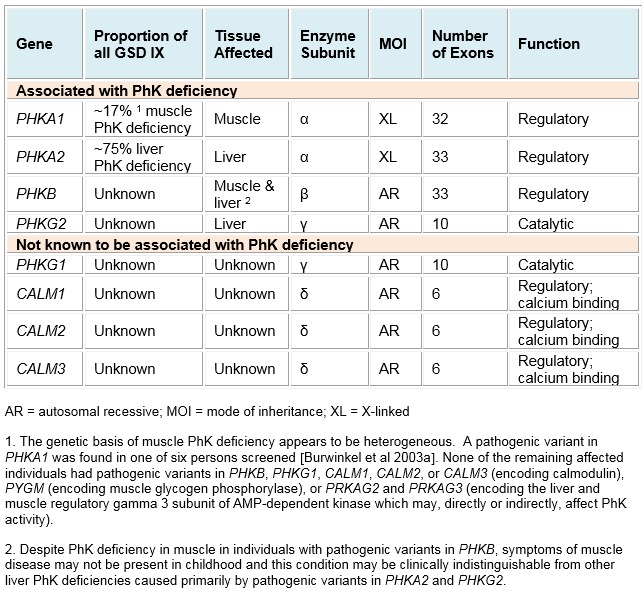
- PHKA1, encoding subunit α, cause the rare X-linked disorder muscle PhK deficiency;
- PHKA2, also encoding subunit α, cause the most common form, liver PhK deficiency (X-linked liver glycogenosis);
- PHKB, encoding subunit β, cause autosomal recessive PhK deficiency in both liver and muscle;
- PHKG2, encoding subunit γ, cause autosomal recessive liver PhK deficiency.
The diagnosis of PhK deficiency is established in a proband with the characteristic clinical findings, a family history of suspected storage disease, and/or a hemizygous pathogenic variant in PHKA1 or PHKA2 or biallelic pathogenic variants in PHKB or PHKG2 identified by molecular genetic testing.
Management.
Treatment of manifestations:
- Liver PhK deficiency. Hypoglycemia can be prevented with frequent daytime feedings that are high in complex carbohydrates and protein. When hypoglycemia or ketosis is present, Polycose® or fruit juice is given orally as tolerated or glucose by IV. Liver manifestations (e.g., cirrhosis, liver failure, portal hypertension) are managed symptomatically.
- Muscle PhK deficiency. Physical therapy based on physical status and function; optimization of blood glucose concentrations by a metabolic nutritionist based on activity.
Surveillance:
- Liver PhK deficiency. Regular evaluation by a metabolic physician and a metabolic nutritionist. Monitoring of blood glucose concentration and blood ketones routinely as well as during times of stress (e.g., illness, intense activity, rapid growth, puberty) and reduced food intake. In children younger than age 18 years, liver ultrasound examination should be performed every 12 to 24 months. With increasing age, CT or MRI using intravenous contrast should be considered to evaluate for complications of liver disease. Echocardiogram should be performed at least every two years.
- Muscle PhK deficiency. Regular evaluation by a metabolic physician, a metabolic nutritionist, and a physical therapist.
Agents/circumstances to avoid:
- Liver PhK deficiency. Large amounts of simple sugars as they will increase liver storage of glycogen; prolonged fasting; high-impact contact sports if significant hepatomegaly is present; drugs known to cause hypoglycemia such as insulin and insulin secretagogues (the sulfonylureas) or drugs known to mask symptoms of hypoglycemia such as beta-blockers; alcohol (which may predispose to hypoglycemia).
- Muscle PhK deficiency. Vigorous exercise; medications like succinylcholine and statins that can cause rhabdomyolysis.
Evaluation of relatives at risk: Molecular genetic testing (if the family-specific pathogenic variant[s] are known) and/or evaluation by a metabolic physician (if the family-specific pathogenic variant[s] are not known) allows early diagnosis and treatment for sibs at increased risk for GSD IX.
Pregnancy management: Individualized dietary management is necessary to maintain euglycemia throughout pregnancy.
Genetic counseling.
PHKA2-related liver PhK deficiency and PHKA1-related muscle PhK deficiency are inherited in an X-linked manner. PHKB-related liver and muscle PhK deficiency and PHKG2-related liver PhK deficiency are inherited in an autosomal recessive manner.
- X-linked inheritance. If the mother of the proband has a pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be heterozygotes (carriers); the development of symptoms in individuals depends on the pattern of X-chromosome inactivation. Affected males pass the pathogenic variant to all of their daughters and none of their sons.
- Autosomal recessive inheritance. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Carrier testing for at-risk relatives, prenatal testing for pregnancies at risk, and preimplantation genetic testing are possible if the pathogenic variant(s) in the family have been identified.
GeneReview Scope
Table
Liver phosphorylase kinase deficiency Muscle phosphorylase kinase deficiency
Diagnosis
Phosphorylase kinase deficiency causing glycogen storage disease type IX (GSD IX) results from deficiency of the enzyme phosphorylase b kinase (PhK), an enzyme with a key regulatory role in the breakdown of glycogen. Deficiency of this enzyme, which is composed of four copies each of four subunits (α, β, γ, and δ), results in considerable clinical variability [Chen 2001, Kishnani & Chen 2010].
For the purposes of this review, phosphorylase kinase (PhK) deficiency has been divided into liver PhK deficiency and muscle PhK deficiency (see Figure 1 and Figure 2). Liver PhK deficiency is further divided into three subtypes based on the gene in which pathogenic variants occur (PHKA2, PHKB, and PHKG2) and inheritance pattern. It should be noted that pathogenic variants in PHKB and, rarely, PHKG2 result in PhK deficiency both in liver and muscle. However, the symptoms from muscle involvement can be mild or absent; thus, this subtype may be clinically indistinguishable from the liver PhK deficiencies caused by pathogenic variants in PHKA2.
Figure 1.
Phosphorylase kinase subunit expression Note: The CALM genes and PHKG1 are not involved in GSD IX.

Figure 2.
Phosphorylase kinase (PhK) enzyme subunits and genes that encode them
PHKA2-related PhK deficiency is also known as X-linked liver glycogenosis (XLG) and is divided into two biochemical subtypes, XLG1 and XLG2, depending on enzyme activity in various tissues.
Muscle PhK deficiency in this review refers to PHKA1-related GSD IX.
PHKG1 has not yet been associated with PhK deficiency.
The delta subunit of PhK, calmodulin, is encoded by three different genes: CALM1, CALM2, and CALM3. To date these have not been associated with PhK deficiency.
Suggestive Findings
Liver or muscle phosphorylase kinase (PhK) deficiency resulting in glycogen storage disease type IX (GSD IX) should be suspected in individuals with the phenotypic findings shown in Table 1.
Table 1.
PhK Deficiency: Suggestive Phenotypic Findings
Establishing the Diagnosis
The diagnosis of PhK deficiency is established in a proband with the characteristic clinical findings (see Suggestive Findings), a family history of suspected storage disease, and/or a hemizygous pathogenic (or likely pathogenic) variant or biallelic pathogenic (or likely pathogenic) variants in one of the genes listed in Table 2. If molecular genetic testing is not diagnostic, PhK activity can be measured in snap-frozen liver biopsy, erythrocytes, leukocytes, and frozen muscle biopsy tissue.
Note: (1) While liver biopsy or muscle biopsy was done routinely in previous years to measure PhK activity, the availability of molecular genetic testing has reduced the need for such invasive procedures. (2) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include any likely pathogenic variants. (3) The identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel, single-gene testing) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of PhK deficiency is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with hepatomegaly and/or muscle weakness or with atypical phenotypic features are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of PhK deficiency, molecular genetic testing approaches can include use of a multigene panel or single-gene testing.
A multigene panel that includes PHKA1, PHKA2, PHKB, PHKG2, and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Note: Single-gene testing may be appropriate given the following clinical presentations:
- For liver PhK deficiency, perform sequence analysis of PHKA2 first, followed by PHKG2, and then PHKB. If only one (in PHKG2 or PHKB) or no pathogenic variant (PHKA2, PHKG2, or PHKB) is found, perform gene-targeted deletion/duplication analysis.
- For muscle PhK deficiency, perform sequence analysis of PHKA1 first. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis.
- In a male with a maternal family history of similarly affected males, it is appropriate to perform sequence analysis of PHKA1 and PHKA2 first depending on muscle/liver symptoms.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by hepatomegaly and/or muscle weakness, or if an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
Exome array (when clinically available) may be considered if exome sequencing is not diagnostic.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in Phosphorylase Kinase Deficiency
Liver Biopsy
Histology
- Histology usually shows distended hepatocytes as a result of excess glycogen accumulation. Bridging portal fibrosis, steatosis, and low-grade inflammatory changes may also be seen [Johnson et al 2012, Tsilianidis et al 2013]. Liver cirrhosis and adenomas have been reported.
- Remarkably elevated glycogen content with normal glycogen structure is found on biochemical testing of snap-frozen liver biopsy tissue.
Enzyme testing. Phosphorylase b kinase (PhK) is reduced in liver, erythrocytes, and leukocytes of most (not all) individuals with liver PhK deficiency.
- Normal PhK activity in erythrocytes is 1.0 μmol/min/g hemoglobin, and in liver it is 0.1 μmol/min/mg protein.
- Abnormal range is <10% of normal level in the tissue being tested.
Note: (1) PhK is a labile enzyme that is highly sensitive to handling conditions and temperature exposure; thus, it is recommended that test blood samples be accompanied by a control blood sample drawn at the same time from an unrelated individual. Samples need to be kept cold (4°C) at all times including during transport. (2) In a subset of affected individuals, in vitro PhK activity is normal or even elevated in erythrocytes and leukocytes and variable in liver.
Muscle Biopsy
Histology
- Excessive amounts of subsarcolemmal glycogen accumulation are found on histology.
- Elevated glycogen content with normal glycogen structure is found on biochemical testing of muscle.
Enzyme testing
- PhK enzyme activity is markedly reduced in muscle but normal in liver, blood cells, and fibroblasts.
- Because PhK enzyme activates the enzyme glycogen phosphorylase in muscle and liver, the activity of glycogen myophosphorylase (phosphorylase-a) may be reduced in muscle in individuals with muscle PhK deficiency.
Clinical Characteristics
Clinical Description
Glycogen storage disease type IX (GSD IX) is caused by PhK deficiency affecting primarily liver or muscle.
Liver PhK Deficiency
While liver PhK deficiency has been considered a mild condition, more severe involvement has been documented [Johnson et al 2012, Tsilianidis et al 2013]. The three subtypes, caused by pathogenic variants in three different genes (PHKA2, PHKB, and PHKG2), cannot be distinguished by their clinical features, which can vary significantly in severity. See also Genotype-Phenotype Correlations.
Presentation. Typically, an affected child presents in the first years of life with hepatomegaly and growth restriction. Hyperketotic hypoglycemia, if present, is usually mild but can be severe and recurrent.
Hepatomegaly
- Hepatomegaly is one of the most common presentations of liver PhK deficiency. The extent of liver enlargement is variable, ranging from mild to massive [Roscher et al 2014].
- Liver fibrosis can occur and in some instances progress to cirrhosis, especially in liver PhK deficiency caused by pathogenic variants in PHKG2; it has also been reported in some individuals with pathogenic variants in PHKA2 [Johnson et al 2012, Tsilianidis et al 2013]. It has not yet been reported in association with PHKB variants but could occur given the findings in other liver PhK deficiency disorders.
- Liver adenoma has been reported but appears to be very rare and mostly associated with the PHKG2-related subtype [Roscher et al 2014, Bali et al 2017].
- Hepatomegaly usually decreases with age. Decrease in liver size and normalization of liver enzymes following treatment with cornstarch and high-protein diet is reported [Beauchamp et al 2007, Roscher et al 2014].
- However, as affected individuals live longer and the natural history and long-term complications are better understood, it is becoming clear that individuals can progress to liver cirrhosis after a period of quiescence and what appears to be normalization.
Growth restriction is most pronounced in childhood, after which catchup growth and normal sexual development occur; most adults reach normal height [Roscher et al 2014, Bali et al 2017].
Hyperketotic hypoglycemia. Hyperketosis, with or without hypoglycemia, can occur following periods of prolonged fasting or decreased nutritional intake, or vomiting and diarrhea during illness. Hyperketonemia is defined as blood 3-β-hydroxybutyrate (βOHB) >1.0 mmol/L (normal <0.3 mmol/L) [Clarke et al 2008].
- Ketotic hypoglycemia varies from occasional to recurrent in some cases [Brown et al 2015, Hodax et al 2017].
- Chronic ketosis indicates poor metabolic control and can affect growth and overall health.
Muscle concerns. Hypotonia and muscle weakness have been observed in some individuals.
- Mild delays in gross motor development are often seen in early childhood.
- Cardiac manifestations are rare; however, asymptomatic interventricular septal hypertrophy was reported in an individual with PHKB-associated liver PhK deficiency [Roscher et al 2014].
Genitourinary findings
- Polycystic ovaries have been noted in females with liver PhK deficiency [Lee & Leonard 1995]. While the frequency of fertility issues has not been well studied, dysmenorrhea, menstrual irregularity, and oligomenorrhea have been reported [Cho et al 2013].
- Renal tubular acidosis has been reported in some individuals [Burwinkel et al 1998a, Beauchamp et al 2007].
Other. Cognitive and/or speech delays that normalized later in life have been reported in a few individuals [Beauchamp et al 2007, Roscher et al 2014].
Adulthood. Symptoms and biochemical abnormalities improve with age in most individuals with liver PhK deficiency [Zhang et al 2017].
Reports of liver cirrhosis and hepatocellular carcinoma show that long-term monitoring is needed in individuals with liver PhK deficiency [Tsilianidis et al 2013, Roscher et al 2014]. Other long-term issues could emerge as affected individuals are followed longitudinally.
Muscle PhK Deficiency
Muscle-specific phosphorylase kinase deficiency is caused by the PHKA1 variant. However, muscle PhK deficiency caused by pathogenic variants in PHKB and (rarely) PHKG2 is also seen.
Presentation. This phenotype can present anytime from childhood to adulthood with a broad range of symptoms including exercise intolerance, muscle cramps, myalgia, myoglobinuria, and progressive muscle weakness [Chen 2001, Kishnani & Chen 2010]. In children, it is primarily manifested as mild gross motor delay [Roscher et al 2014, Bali et al 2017].
- Minor muscle involvement has been reported in some affected individuals, particularly associated with PHKG2-related muscle PhK deficiency.
- Interventricular septal hypertrophy has been reported in an individual with pathogenic variants in PHKB [Roscher et al 2014].
Muscle concerns can include the following:
- Exercise-induced cramps, muscle pain, and fatigue [Wuyts et al 2005, Preisler et al 2012]
- Proximal limb-girdle weakness, especially of the pelvic girdle [Wuyts et al 2005]
- Progressive muscle weakness leading to muscular atrophy [Burwinkel et al 2000]
- Rhabdomyolysis [Burwinkel et al 2000]
- Asymptomatic elevation of plasma CK in some individuals
Other findings
- Liver involvement
- Liver involvement has not been reported in GSD IX caused by pathogenic variants in PHKA1.
- Hepatomegaly and hypoglycemia are present in some individuals with pathogenic variants in PHKB.
- Interventricular septal hypertrophy has been reported in an individual with a PHKB variant [Roscher et al 2014].
- One adult male with asymptomatic myopathy and cognitive impairment has been reported, suggesting wide variability in the clinical findings associated with pathogenic variants in PHKA1 [Echaniz-Laguna et al 2010]. However, it is possible that another cause exists for the cognitive impairment in this person.
Genotype-Phenotype Correlations
Pathogenic variants in PHKA1 result in muscle glycogenosis; pathogenic variants in PHKA2 and PHKG2 cause liver glycogenosis; pathogenic variants in PHKB and, rarely, PHKG2 cause liver and muscle glycogenosis (muscle signs are variably present).
There is no consistent genotype-phenotype correlation for pathogenic variants in any of the four genes. Pathogenic variants in PHKG2 appear to result in more severe disease with an increased risk of liver fibrosis and cirrhosis, although persons with a milder course have been observed. Progressive liver disease has also been noted in individuals with pathogenic variants in PHKA2.
Nomenclature
Liver PhK deficiency. Historically, the numeric classification of liver PhK deficiency has ranged from GSD type VIa and VIb to GSD VIII to GSD IX.
Note: (1) Deficiency of the enzyme glycogen phosphorylase that causes GSD V (muscle specific) or GSD VI (liver specific) is distinct from deficiency of the enzyme PhK that causes GSD IX. However, confusion may have arisen in the past reclassification of these types of GSD: because the enzyme PhK activates the enzyme glycogen phosphorylase, PhK deficiency can also result in phosphorylase deficiency. (2) The classification GSD VIII no longer exists: in the past GSD VIII was used to describe some cases of PhK deficiency.
Liver PhK deficiency has been further subclassified into:
- GSD IXa, now known as PHKA2-related glycogen storage disease type IX;
- GSD IXb, now known as PHKB-related glycogen storage disease type IX;
- GSD IXc, now known as PHKG2-related glycogen storage disease type IX.
Muscle PhK deficiency has been called GSD Vb and GSD IXd.
Prevalence
Liver PhK deficiency is thought to account for about 25% of all GSDs with an estimated prevalence of 1:100,000 [Maichele et al 1996]. However, the disorder may be underdiagnosed as a result of the variable presentation and challenges with diagnostic confirmation.
Muscle PhK deficiency appears to be rare, but could be underdiagnosed because of the milder and more variable muscle symptoms.
No populations are known to have an increased prevalence of PhK deficiency.
Genetically Related (Allelic) Disorders
No other phenotypes are known to be associated with pathogenic variants in PHKA1, PHKA2, PHKB, or PHKG2.
Differential Diagnosis
Table 3.
Genetic Disorders to Consider in the Differential Diagnosis of Phosphorylase Kinase (PhK) Deficiency
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with liver PhK deficiency, the following evaluations are recommended if they have not already been completed:
- Measurement of blood glucose concentration (normal >70 mg/dL) for two to three days:
- Upon waking in the morning;
- Prior to meals and nighttime supplementation with oral cornstarch; and
- After activity
- Measurement of blood ketone levels for two to three days:
- Upon waking in the morning;
- Prior to meals and nighttime supplementation with oral cornstarch; and
- After activity.
Elevated blood ketones (beta-hydroxybutyrate >1.0 mmol/L) could be an indicator of suboptimal metabolic control and pending hypoglycemia. - Liver imaging, if not performed in the past year. The type of liver imaging (ultrasound, MRI, or CT) is determined by factors such as age and underlying liver status (e.g., liver cirrhosis).
- Basic metabolic panel including liver enzymes (AST, ALT, and alkaline phosphatase)
- Prothrombin time
- Lipid panel (cholesterol and triglyceride concentrations)
- Serum creatine kinase measurement (Some individuals with liver PhK deficiency have muscle involvement.)
- Baseline echocardiogram (Baseline echocardiogram may be done as a precaution as interventricular septal hypertrophy was found in an individual with GSD IX caused by pathogenic variants in PHKB [Roscher et al 2014].)
To establish the extent of disease and needs of an individual diagnosed with muscle PhK deficiency, the following evaluations are suggested:
- Physical therapy evaluation
- Serum creatine kinase measurement
Consultation with a clinical geneticist and/or genetic counselor is recommended for both PhK phenotypes.
Treatment of Manifestations
Liver PhK Deficiency
Goal of treatment is to keep blood glucose between 70 and 100 mg/dL. The target range for blood βOHB is 0.0-0.2 mmol/L.
Hypoglycemia can be prevented with frequent daytime feedings that are high in complex carbohydrates and protein.
- The dose of cornstarch can range from 0.6 to 2.5 g/kg every six hours based on clinical symptoms. A waxy maize extended-release cornstarch, Glycosade®, allows sustained release of glucose, especially overnight in some cases.
- Protein should be given as 15% to 25% of total calories, 2-3 g protein/kg body weight/day (tailored to the individual's age) as long as renal function is normal. Protein provides an alternative source of glucose via intact gluconeogenesis.
- Fats should provide ~30% of total calories. Saturated fats should be restricted to <10% of total calories and cholesterol to <300 mg/day.
- Overtreatment with cornstarch should be avoided as it can cause diarrhea, weight gain, and insulin resistance.
For signs of hypoglycemia or ketosis, Polycose® or fruit juice should be given orally (if oral intake is tolerated) followed by a snack high in complex carbohydrates and protein. Blood glucose and ketone concentrations should be monitored periodically to ensure that they return to normal. If oral intake is not tolerated, an IV should be started.
When intravenous dextrose support is required, a concentration of 10% dextrose should be used at a rate that is 1-1.25x the maintenance rate with appropriate electrolytes. The rate can be increased based on blood glucose levels. Fluids with less concentrated dextrose (i.e., 5% dextrose) could result in fluid overload at the rate required to maintain blood glucose above 70 mg/dL and prevent ketosis.
Symptomatic management of liver manifestations such as cirrhosis, liver failure, and portal hypertension is appropriate.
Muscle PhK Deficiency
Signs and symptoms should be managed as with other muscle GSDs, such as GSD III [Kishnani et al 2010]:
- Physical therapy evaluation and intervention based on physical status and function
- Coordination with a metabolic nutritionist regarding monitoring and optimizing of blood glucose concentrations based on levels of exercise and activity
Prevention of Secondary Complications
Liver PhK Deficiency
General medical care. During childhood, routine immunizations should be given on the recommended schedule. Any immunizations that may prevent illness, such as influenza leading to hypoglycemia, should be offered.
Surgery/anesthesia. If prolonged fasting is required in preparation for surgery, the individual should be admitted overnight for IV dextrose support to maintain blood glucose concentration >70 mg/dL and to prevent ketosis.
Perioperative care for elective procedures should include IV dextrose infusion preoperatively, which should start as soon as the individual is made NPO with a goal of maintaining blood glucose levels >70 mg/dL and to prevent ketosis. A concentration of 10% dextrose should be used at 1.25-1.5x the maintenance rate. Using a lower concentration of dextrose such as 5% dextrose can result in fluid overload. Blood glucose and βOHB levels should be measured upon arrival and hourly. Continue with intraoperative and postoperative IV dextrose infusion to prevent hypoglycemia. IV dextrose should be tapered off gradually over two to three hours as the individual tolerates the usual diet. Abrupt discontinuation of fluids could result in hypoglycemia.
In individuals with liver fibrosis or cirrhosis, anesthetic agents with hepatic side effects should be avoided.
If general anesthesia is required, malignant hyperthermia precautions should be taken as individuals with liver PhK deficiency may have increased CK levels and myopathy [Author, personal experience] (see Malignant Hyperthermia Susceptibility).
Physical activity. Contact sports should be avoided in individuals with hepatomegaly. Blood glucose and ketones should be monitored during and after exercise, based on the needs of the individual. This is because of the extreme clinical heterogeneity in those with GSD IX.
Muscle PhK Deficiency
Lipid-lowering drugs (e.g., statins) that can worsen or unmask myopathy should be used cautiously due to the risk of rhabdomyolysis.
If general anesthesia is required, malignant hyperthermia precautions should be taken as individuals with muscle PhK deficiency have increased CK levels and myopathy (see Malignant Hyperthermia Susceptibility).
During childhood, routine immunizations should be given on the recommended schedule. Hepatitis B vaccine should be administered when liver enzymes of the individual are within normal limits.
Surveillance
Liver PhK deficiency
- Regular evaluation by a metabolic physician familiar with liver PhK deficiency to monitor medical issues and a metabolic nutritionist to give dietary recommendations and monitor cornstarch requirement
- Regular monitoring of blood glucose concentration and ketones, as recommended by a metabolic physician and nutritionist. Blood glucose concentrations and ketones should also be measured during times of stress including illness, intense activity, rapid growth, puberty, and pregnancy; and at any time in which intake of food is reduced, or meal and/or cornstarch dose or scheduling is altered.Note: It is possible that blood glucose concentrations may be normal when moderate to large ketosis in liver PhK deficiency results from increased fatty acid oxidation and upregulated gluconeogenesis. The role of ketone monitoring in this setting as a marker of metabolic control requires further systematic investigation.
- Liver imaging. In children younger than age 18 years, liver ultrasound examination every 12 to 24 months. With increasing age, consideration of CT or MRI using intravenous contrast to evaluate for complications of liver disease such as liver adenomas and cirrhosis
- Follow-up echocardiogram. No guidelines established; follow up approximately every two years or earlier if symptoms are present
Muscle PhK deficiency
- Regular evaluation by a metabolic physician familiar with liver PhK deficiency to monitor medical issues and a metabolic nutritionist to give dietary recommendations and monitor cornstarch requirement
- Regular evaluation by a physical therapist to look for progression in symptoms and to guide exercise program
Agents/Circumstances to Avoid
Liver PhK deficiency. Affected Individuals should avoid the following:
- Large amounts of simple sugars, as they will increase liver storage of glycogen and may result in rapid fluctuations in levels of blood glucose and insulin
- Prolonged fasting
- High-impact contact sports if significant (moderate to massive) hepatomegaly is present. The final decision is based on clinician judgment.
- Drugs known to:
- Cause hypoglycemia, such as insulin and insulin secretagogues (the sulfonylureas)
- Mask symptoms of hypoglycemia, such as beta-blockers
- Alcohol, as this may predispose to hypoglycemia
- Glucagon, as glycogenolysis is defective
- Augmentin®, as it can cause malabsorption and contains clavulanic acid, which is associated with idiopathic liver disease
- Growth hormone therapy unless there is proven growth hormone deficiency, as it can promote ketosis and development of liver adenomas
Hypoglycemic events in adults with liver PhK deficiency are relatively uncommon; however, caution should be used with drugs causing potential hypoglycemia, particularly in persons with impaired liver function.
Muscle PhK deficiency. Affected individuals should avoid the following:
- Vigorous exercise
- Medications that can cause rhabdomyolysis (e.g., succinylcholine)
- Statins (to be used with caution, as they can cause rhabdomyolysis)
Evaluation of Relatives at Risk
Molecular genetic testing (if the family-specific variant[s] are known) and/or evaluation by a metabolic physician during the first year of life (if the family-specific variant[s] are not known) allows for early diagnosis and treatment for sibs at increased risk for GSD IX.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Obstetric/Gynecologic Care
Females with GSD IX should be evaluated for symptoms for polycystic ovary syndrome.
Pregnancy Management
Ideally, females with PhK deficiency should consult with their health care team and maintain optimal metabolic control before conception.
It is extremely important that euglycemia be maintained throughout pregnancy to avoid upregulation of counter-regulatory hormones, which would result in lipolysis and ketosis, with risk of fetal demise. The appropriate diet during pregnancy is unique to each individual. For some, this may only require following a regular healthy diet, but for many it may mean increasing snacks to include more complex carbohydrates and protein and/or adding or increasing the amount of cornstarch. Blood glucose concentrations and ketones should also be measured during pregnancy on a regular basis to ensure euglycemia. Adequate amounts of protein are necessary to provide an alternate source of glucose via gluconeogenesis.
Therapies under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
PHKA2-related liver PhK deficiency and PHKA1-related muscle PhK deficiency are inherited in an X-linked manner.
PHKB-related liver and muscle PhK deficiency and PHKG2-related liver PhK deficiency are inherited in an autosomal recessive manner.
X-Linked Inheritance – Risk To Family Members
Parents of a male proband
- The father of an affected male will not have the disorder nor will he be hemizygous for a PHKA1 or PHKA2 pathogenic variant.
- In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote (carrier). If a woman has more than one affected child and no other affected relatives, and if the PHKA1 or PHKA2 pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism (maternal germline mosaicism has been reported in PHKA2-related liver PhK deficiency) [Qin et a 2016].
- If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote (carrier), or the affected male may have a de novo pathogenic variant, in which case the mother is not a carrier. To date, PHKA2-related liver PhK deficiency caused by a de novo pathogenic variant has been documented in one individual [Rodríguez-Jiménez et al 2017]. The overall frequency of de novo pathogenic variants in PHKA1 and PHKA2 is unknown.
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
- If the mother of the proband has a PHKA1 or PHKA2 pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the variant will be heterozygotes (carriers).
- PHKA1. No symptoms have been reported in females who are heterozygous for a PHKA1 pathogenic variant, although development of symptoms may occur, in theory, if skewed X-chromosome inactivation is present.
- PHKA2. Females who are heterozygous for a PHKA2 pathogenic variant may be unaffected, have mild hepatomegaly, or (rarely) have more severe symptoms depending on the pattern of X-chromosome inactivation [Author, personal observation].
- If the affected male represents a simplex case (i.e., a single occurrence in a family) and if the PHKA1 or PHKA2 pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is slightly greater than that of the general population (though still <1%) because of the theoretic possibility of maternal germline mosaicism.
Offspring of a male proband. Affected males transmit the pathogenic variant to all of their daughters and none of their sons.
Other family members. An affected male's maternal aunts may be at risk of being carriers, and the aunts' offspring, depending on their sex, may be at risk of being carriers or of being affected.
Note: Molecular genetic testing may be able to identify the family member in whom a de novo pathogenic variant arose, information that could help determine genetic risk status of the extended family.
Heterozygote detection. Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the pathogenic variant in the family has been identified in the proband.
Note: (1) Identification of female carriers requires either (a) prior identification of the PHKA1 or PHKA2 pathogenic variant in the family or, (b) if an affected male is not available for testing, molecular genetic testing first by sequence analysis, and then, if no variant is identified, using deletion/duplication analysis if available. (2) Enzyme-based carrier testing is not recommended for determining carrier status. (3) Female carriers are heterozygotes for these X-linked disorders. Females who are heterozygous for a PHKA2 pathogenic variant may develop mild hepatomegaly, short stature in childhood, and biochemical abnormalities [Willems et al 1990, Morava et al 2005], and theoretically more severe symptoms (including hypoglycemia) resulting from skewed X-chromosome inactivation. No symptoms have been reported in females who are heterozygous for a pathogenic variant in PHKA1 [Bak et al 2001], but it is possible that they, too, could exhibit symptoms as a result of skewed X-chromosome inactivation.
Autosomal Recessive Inheritance – Risk To Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one PHKB or PHKG2 pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing PhK deficiency.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing PhK deficiency.
Offspring of a proband. The offspring of an individual with autosomal recessive PhK deficiency are obligate heterozygotes (carriers) for a pathogenic variant in PHKB or PHKG2.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier.
Carrier detection. Carrier testing for relatives at risk for the two subtypes of autosomal recessive PhK deficiency (caused by pathogenic variants in PHKB or PHKG2) requires prior identification of the pathogenic variants in the family.
Enzyme-based carrier testing is not recommended for determining carrier status.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk of being carriers or of being affected.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing for PhK deficiency are possible. Molecular testing is the preferred method for prenatal testing.
Biochemical testing. Prenatal testing based on PK enzyme testing is not available and not recommended. PK is a very labile kinase enzyme so that any prenatal testing or carrier testing results may not be reliable.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While use of prenatal testing is a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Association for Glycogen Storage Disease
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Phosphorylase Kinase Deficiency: Genes and Databases
Table B.
OMIM Entries for Phosphorylase Kinase Deficiency (View All in OMIM)
Molecular Pathogenesis
The enzyme phosphorylase kinase (PhK) activates liver glycogen phosphorylase and muscle glycogen phosphorylase in response to neuronal and hormonal stimuli and thus is a key regulatory enzyme in glycogen breakdown. In liver PhK deficiency, the inability to break down glycogen results in risk for hypoglycemia and glycogen accumulation in the liver, which in turn causes hepatomegaly and liver damage.
PhK is a multi-subunit enzyme composed of four copies each of four subunits (α, β, γ, and δ). The gamma (γ) subunit contains the catalytic activity and is regulated by the alpha (α), beta (β), and delta (δ) subunits. The inhibitory effect of the alpha and beta subunits is modulated by phosphorylation (phosphorylation removes the inhibitory effect); calcium levels modulate the regulatory effect of the delta subunit (calmodulin).
Each PhK subunit is encoded by at least one gene: PHKA1 and PHKA2 encode the muscle and liver isoforms of the α-subunit, respectively; PHKB encodes the liver and muscle β-subunits; PHKG1 and PHKG2 encode the muscle and liver isoforms of the γ-subunit, respectively; and the δ-subunit, calmodulin, is encoded by three genes: CALM1, CALM2, and CALM3. Expression of these genes is tissue dependent (Figure 1). Further complexity is introduced by tissue-specific alternative splicing. The complexity of the enzyme PhK explains to some degree the clinical and biochemical heterogeneity of PhK deficiency.
Of these eight genes, four are known to contain pathogenic variants that cause PhK enzyme deficiency (Figure 2).
The two X-linked genes are:
- PHKA1, which causes the rare disorder X-linked muscle PhK deficiency; and
- PHKA2, which causes the most common form of liver PhK deficiency, also known as X-linked liver glycogenosis (XLG).
The two autosomal genes are:
- PHKB, which causes PhK deficiency in both liver and muscle but manifests primarily with liver symptoms with or without muscle involvement; and
- PHKG2, which causes PhK deficiency in liver.
Muscle PhK activity is normal in individuals with pathogenic variants in either PHKA2 or PHKG2 and can be deficient in those with pathogenic variants in PHKB.
PHKA1
Gene structure. Alternatively spliced transcript variants encoding different isoforms have been identified in this gene. The longest transcript (NM_002637.3) consists of 32 exons and is transcribed into a 6-kb cDNA. PHKA1 spans approximately 133 kb of genomic DNA. A pseudogene, PHKA1P1, has been found on chromosome 1p22.2. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. To date, seven pathogenic variants have been reported in PHKA1, each of which was found in only one or two individuals. Pathogenic variants include missense (3), frameshift (2), nonsense(1), and splice site changes (1) [Wehner et al 1994, Bruno et al 1998, Burwinkel et al 2003a, Wuyts et al 2005, Ørngreen et al 2008, Echaniz-Laguna et al 2010].
A frameshift variant in mouse ortholog Phka1 causes PhK deficiency in the I-strain mouse [Schneider et al 1993].
Normal gene product. PHKA1 encodes the muscle isoform of the alpha subunit of PhK, a 1,223-amino-acid protein (NP_002628.2).
Two alternatively spliced transcript variants encoding different isoforms have been identified [Harmann et al 1991]; alpha-FM is the predominant form in fast-twitch skeletal muscle and is also expressed in brain, while alpha-prime is the predominant form in slow-twitch skeletal muscle. Alpha-prime has an internal deletion of 59 amino acids (amino acids 654-712) when compared to alpha-FM.
The degree of phosphorylation of the alpha subunit regulates the activity of PhK; the greater the phosphorylation, the less the inhibitory effect.
Abnormal gene product. Muscle PhK deficiency due to pathogenic variants in PHKA1 appears to cause myopathy through a defect in glycogen availability during submaximal exercise (oxidative metabolism; e.g., cycle test) presumably because PhK is required to activate glycogen phosphorylase under these conditions. Interestingly, anaerobic glycogenolysis is normal, suggesting that other regulatory factors are involved in phosphorylase activation in this situation [Ørngreen et al 2008].
Complete lack of PHKA1 protein is predicted to affect formation or stability of PhK holoenzyme. Production of an altered PHKA1 protein, resulting from missense variants, may affect its ability to interact with other subunits or to activate PhK activity.
PHKA2
Gene structure. The gene contains 33 exons [Hendrickx et al 1999] and spans 91.3 kb of DNA. Alternatively spliced transcript variants have been reported, but the full-length nature of these variants has not been determined. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. More than 100 different pathogenic variants have been reported in PHKA2. Most of them are missense or nonsense variants or small deletions causing frameshifts. Only seven gross deletions/duplications have been reported. The pathogenic variants are distributed throughout the gene.
Table 4.
PHKA2 Pathogenic Variants Discussed in This GeneReview
Normal gene product. PHKA2 encodes the liver alpha subunit of PhK. A 5,325-bp mRNA is translated into a 1,235-amino-acid protein with high expression in liver and brain, but not in muscle [Hendrickx et al 1993]. PHKA2 is highly homologous to PHKA1 and PHKB.
Abnormal gene product. Two biochemical subtypes of X-linked glycogenosis (XLG) are caused by pathogenic variants in PHKA2 [Hendrickx et al 1994, Burwinkel et al 1996, Hendrickx et al 1996, Burwinkel et al 1998a, Hendrickx et al 1999]:
- XLG1, the more common form, in which in vitro PhK activity is deficient in peripheral blood cells and liver
- XLG2, in which in vitro PhK activity in peripheral blood cells is normal or even elevated and activity in liver is variable
While not yet fully understood, there are various theories as to how different pathogenic variants in PHKA2 could result in these different biochemical subtypes.
Regarding correlation between genotype and biochemical phenotype, Hendrickx et al [1999] suggested the following:
- PHKA2 variants resulting in reduced amounts of alpha subunit protein (e.g., nonsense and frameshift variants or missense variants that destabilize the protein) cause detectable PhK deficiency in vitro (XLG1 biochemical subtype).
- PHKA2 variants that disrupt activation of PhK enzyme activity (e.g., missense variants or small in-frame insertions or deletions affecting regulatory sites of the enzyme) can result in the normal PhK activity that is observed in vitro in some affected persons (XLG2 biochemical subtype).
These subtle changes may allow normal amounts of PhK to be made but affect enzyme function [Maire et al 1991, Hendrickx et al 1994, Burwinkel et al 1996, Hendrickx et al 1996, Burwinkel et al 1997b, Burwinkel et al 1998a, Hendrickx et al 1998].
Carrière et al [2008] showed that PHKA2 missense variants and small in-frame deletions/insertions are concentrated into two domains of the protein, the N-terminal glucoamylase domain (principally leading to XLG2) and the C-terminal calcineurin B-like domain (domain D; principally leading to XLG1).
Further studies are needed to determine the molecular basis of the XLG1 and XLG2 biochemical subtypes. Of note, the same PHKA2 variant (p.Arg295His) has been associated with normal and deficient PhK activity in vitro, suggesting that other factors, such as handling of the specimen and laboratory methodologies, can also affect the biochemical phenotype [Hendrickx et al 1999].
PHKB
Gene structure. Alternatively spliced transcript variants encoding different isoforms have been identified in this gene. The longer transcript variant, NM_000293.2, is composed of 33 exons spanning 239 kb of genomic DNA. Exons 26 and 27 are two homologous, mutually exclusively spliced exons that encode muscle and non-muscle PHKB, respectively; exon 2 is a facultatively used cassette exon encoding an alternative N-terminus [Wüllrich-Schmoll & Kilimann 1996]. For a detailed summary of gene and protein information, see Table A, Gene.
Two processed pseudogenes have been identified: PHKBP1 on chromosome 20p12.3-20p12.2 and PHKBP2 on chromosome 14q13.3 [Wüllrich-Schmoll & Kilimann 1996].
Pathogenic variants. More than 20 variants suspected or known to be pathogenic have been reported in PHKB. These include nonsense, missense, splice site, frameshift, and gross deletion changes [Burwinkel et al 1997b, van den Berg et al 1997, Burwinkel et al 2003a, Beauchamp et al 2007]. Two of the missense changes, p.Met185Ile and p.Gln657Lys, were identified in heterozygotes in whom no other pathogenic variant was identified, and thus their significance is unknown [Burwinkel et al 1997a, Burwinkel et al 2003a, Beauchamp et al 2007].
Table 5.
PHKB Pathogenic Variants Discussed in This GeneReview
Normal gene product. PHKB encodes the beta subunit of liver or muscle PhK. The degree of phosphorylation of the beta subunit determines the activity of the enzyme PhK.
Abnormal gene product. It is not known exactly how PHKB pathogenic variants result in PhK deficiency. Lack of PHKB protein would affect formation of the normal PhK holoenzyme, and an abnormal PHKB protein would presumably affect its interaction with other PhK subunits and its regulatory function. Biochemical evidence suggests that an alpha-gamma-delta complex may form in the absence of the beta subunit, explaining the residual enzyme activity seen in some individuals and the mild clinical features [Burwinkel et al 1997a, Brushia & Walsh 1999].
PHKG2
Gene structure. Alternatively spliced transcript variants encoding different isoforms have been identified in this gene. The PHKG2 longer transcript isoform NM_000294.2 comprises ten exons spanning 9 kb of genomic DNA. A complex microsatellite repeat has been identified at the beginning of intron 2 [Burwinkel et al 1998b]. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. More than 30 pathogenic variants have been reported in PHKG2 including missense, nonsense, splice site, and frameshift changes [Maichele et al 1996, van Beurden et al 1997, Burwinkel et al 1998b, Burwinkel et al 2000, Burwinkel et al 2003b, Beauchamp et al 2007].
Normal gene product. PHKG2 encodes the catalytic gamma subunit of liver PhK, a 406-amino-acid protein, NP_000285.1. Alternative splicing creates a variant of 374 amino acids with a different C-terminus.
Abnormal gene product. Pathogenic variants in PHKG2 are expected to affect the catalytic ability of the gamma subunit either by resulting in production of no protein or affecting the stability or confirmation of the protein.
Chapter Notes
Acknowledgments
We would like to thank Dr YT Chen, Denise Petersen, and Keri Fredrickson for their useful discussions and professional contributions to increasing the understanding and knowledge of this complicated disorder.
Revision History
- 1 November 2018 (ha) Comprehensive update posted live
- 31 May 2011 (me) Review posted live
- 7 September 2010 (db) Original submission
References
Literature Cited
- Alfadhel M, Benmeakel M, Hossain MA, Al Mutairi F, Al Othaim A, Alfares AA, Al Balwi M, Alzaben A, Eyaid W. Thirteen year retrospective review of the spectrum of inborn errors of metabolism presenting in a tertiary center in Saudi Arabia. Orphanet J Rare Dis. 2016;11:126. [PMC free article: PMC5024448] [PubMed: 27629047]
- Bak H, Cordato D, Carey WF, Milder D. Adult-onset exercise intolerance due to phosphorylase b kinase deficiency. J Clin Neurosci. 2001;8:286–7. [PubMed: 11386811]
- Bali DS, Goldstein JL, Fredrickson K, Austin S, Pendyal S, Rehder C, Kishnani PS. Clinical and molecular variability in patients with PHKA2 variants and liver phosphorylase b kinase deficiency. JIMD Rep. 2017;37:63–72. [PMC free article: PMC5740042] [PubMed: 28283841]
- Bali DS, Goldstein JL, Fredrickson K, Rehder C, Boney A, Austin S, Weinstein DA, Lutz R, Boneh A, Kishnani PS. Variability of disease spectrum in children with liver phosphorylase kinase deficiency caused by mutations in the PHKG2 gene. Mol Genet Metab. 2014;111:309–13. [PMC free article: PMC3952947] [PubMed: 24389071]
- Beauchamp NJ, Dalton A, Ramaswami U, Niinikoski H, Mention K, Kenny P, Kolho KL, Raiman J, Walter J, Treacy E, Tanner S, Sharrard M. Glycogen storage disease type IX: high variability in clinical phenotype. Mol Genet Metab. 2007;92:88–99. [PubMed: 17689125]
- Brown LM, Corrado MM, van der Ende RM, Derks TG, Chen MA, Siegel S, Hoyt K, Correia CE, Lumpkin C, Flanagan TB, Carreras CT, Weinstein DA. Evaluation of glycogen storage disease as a cause of ketotic hypoglycemia in children. J Inherit Metab Dis. 2015;38:489–93. [PubMed: 25070466]
- Bruno C, Manfredi G, Andreu AL, Shanske S, Krishna S, Ilse WK, DiMauro S. A splice junction mutation in the alpha(M) gene of phosphorylase kinase in a patient with myopathy. Biochem Biophys Res Commun. 1998;249:648–51. [PubMed: 9731190]
- Brushia RJ, Walsh DA. Phosphorylase kinase: the complexity of its regulation is reflected in the complexity of its structure. Front Biosci. 1999;4:D618–41. [PubMed: 10487978]
- Burwinkel B, Amat L, Gray RG, Matsuo N, Muroya K, Narisawa K, Sokol RJ, Vilaseca MA, Kilimann MW. Variability of biochemical and clinical phenotype in X-linked liver glycogenosis with mutations in the phosphorylase kinase PHKA2 gene. Hum Genet. 1998a;102:423–9. [PubMed: 9600238]
- Burwinkel B, Hu B, Schroers A, Clemens PR, Moses SW, Shin YS, Pongratz D, Vorgerd M, Kilimann MW. Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet. 2003a;11:516–26. [PubMed: 12825073]
- Burwinkel B, Maichele AJ, Aagenaes O, Bakker HD, Lerner A, Shin YS, Strachan JA, Kilimann MW. Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB). Hum Mol Genet. 1997a;6:1109–15. [PubMed: 9215682]
- Burwinkel B, Moses SW, Kilimann MW. Phosphorylase-kinase-deficient liver glycogenosis with an unusual biochemical phenotype in blood cells associated with a missense mutation in the beta subunit gene (PHKB). Hum Genet. 1997b;101:170–4. [PubMed: 9402963]
- Burwinkel B, Rootwelt T, Kvittingen EA, Chakraborty PK, Kilimann MW. Severe phenotype of phosphorylase kinase-deficient liver glycogenosis with mutations in the PHKG2 gene. Pediatr Res. 2003b;54:834–9. [PubMed: 12930917]
- Burwinkel B, Shin YS, Bakker HD, Deutsch J, Lozano MJ, Maire I, Kilimann MW. Mutation hotspots in the PHKA2 gene in X-linked liver glycogenosis due to phosphorylase kinase deficiency with atypical activity in blood cells (XLG2). Hum Mol Genet. 1996;5:653–8. [PubMed: 8733134]
- Burwinkel B, Shiomi S, Al Zaben A, Kilimann MW. Liver glycogenosis due to phosphorylase kinase deficiency: PHKG2 gene structure and mutations associated with cirrhosis. Hum Mol Genet. 1998b;7:149–54. [PubMed: 9384616]
- Burwinkel B, Tanner MS, Kilimann MW. Phosphorylase kinase deficient liver glycogenosis: progression to cirrhosis in infancy associated with PHKG2 mutations (H144Y and L225R). J Med Genet. 2000;37:376–7. [PMC free article: PMC1734590] [PubMed: 10905889]
- Carrière C, Jonic S, Mornon JP, Callebaut I. 3D mapping of glycogenosis-causing mutations in the large regulatory alpha subunit of phosphorylase kinase. Biochim Biophys Acta. 2008;1782:664–70. [PubMed: 18950708]
- Clarke W, Jones T, Rewers A, Dunger D, Klingensmith GJ. Assessment and management of hypoglycemia in children and adolescents with diabetes. Pediatr Diabetes. 2008;9:165–74. [PubMed: 18416698]
- Chen YT. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. Chapter 71. New York: McGraw-Hill; 2001:1521-51.
- Cho SY, Lam CW, Tong SF, Siu WK. X-linked glycogen storage disease IXa manifested in a female carrier due to skewed X chromosome inactivation. Clin Chim Acta. 2013;426:75–8. [PubMed: 24055370]
- Choi R, Park HD, Kang B, Choi SY, Ki CS, Lee SY, Kim JW, Song J, Choe YH. PHKA2 mutation spectrum in Korean patients with glycogen storage disease type IX: prevalence of deletion mutations. BMC Med Genet. 2016;17:33. [PMC free article: PMC4839068] [PubMed: 27103379]
- Davit-Spraul A, Piraud M, Dobbelaere D, Valayannopoulos V, Labrune P, Habes D, Bernard O, Jacquemin E, Baussan C. Liver glycogen storage diseases due to phosphorylase system deficiencies: diagnosis thanks to non invasive blood enzymatic and molecular studies. Mol Genet Metab. 2011;104:137–43. [PubMed: 21646031]
- Echaniz-Laguna A, Akman HO, Mohr M, Tranchant C, Talmant-Verbist V, Rolland MO, Dimauro S. Muscle phosphorylase b kinase deficiency revisited. Neuromuscul Disord. 2010;20:125–7. [PubMed: 20080404]
- Harmann B, Zander NF, Kilimann MW. Isoform diversity of phosphorylase kinase alpha and beta subunits generated by alternative RNA splicing. J Biol Chem. 1991;266:15631–7. [PubMed: 1874721]
- Hendrickx J, Bosshard NU, Willems P, Gitzelmann R. Clinical, biochemical and molecular findings in a patient with X-linked liver glycogenosis followed for 40 years. Eur J Pediatr. 1998;157:919–23. [PubMed: 9835437]
- Hendrickx J, Coucke P, Bossuyt P, Wauters J, Raeymaekers P, Marchau F, Smit GP, Stolte I, Sardharwalla IB, Berthelot J, et al. X-linked liver glycogenosis: localization and isolation of a candidate gene. Hum Mol Genet. 1993;2:583–9. [PubMed: 8518797]
- Hendrickx J, Coucke P, Hors-Cayla MC, Smit GP, Shin YS, Deutsch J, Smeitink J, Berger R, Lee P, Fernandes J. Localization of a new type of X-linked liver glycogenosis to the chromosomal region Xp22 containing the liver alpha-subunit of phosphorylase kinase (PHKA2). Genomics. 1994;21:620–5. [PubMed: 7959740]
- Hendrickx J, Dams E, Coucke P, Lee P, Fernandes J, Willems PJ. X-linked liver glycogenosis type II (XLG II) is caused by mutations in PHKA2, the gene encoding the liver alpha subunit of phosphorylase kinase. Hum Mol Genet. 1996;5:649–52. [PubMed: 8733133]
- Hendrickx J, Lee P, Keating JP, Carton D, Sardharwalla IB, Tuchman M, Baussan C, Willems PJ. Complete genomic structure and mutational spectrum of PHKA2 in patients with x-linked liver glycogenosis type I and II. Am J Hum Genet. 1999;64:1541–9. [PMC free article: PMC1377897] [PubMed: 10330341]
- Hodax JK, Uysal S, Quintos JB, Phornphutkul C. Glycogen storage disease type IX and growth hormone deficiency presenting as severe ketotic hypoglycemia. J Pediatr Endocrinol Metab. 2017;30:247–51. [PubMed: 28085675]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Johnson AO, Goldstein JL, Bali D. Glycogen storage disease type IX: novel PHKA2 missense mutation and cirrhosis. J Pediatr Gastroenterol Nutr. 2012;55:90–2. [PubMed: 21857251]
- Kishnani P, Chen YT. Defects in metabolism of carbohydrates. In: Kliegman RM, Stanton BMD, St. Geme J, Schor N, Behrman RE, eds. Nelson Textbook of Pediatrics. 19 ed. Amsterdam: Elsevier; 2010.
- Kishnani PS, Austin SL, Arn P, Bali DS, Boney A, Case LE, Chung WK, Desai DM, El-Gharbawy A, Haller R, Smit GP, Smith AD, Hobson-Webb LD, Wechsler SB, Weinstein DA, Watson MS. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12:446–63. [PubMed: 20631546]
- Lee PJ, Leonard JV. The hepatic glycogen storage diseases--problems beyond childhood. J Inherit Metab Dis. 1995;18:462–72. [PubMed: 7494404]
- Maichele AJ, Burwinkel B, Maire I, Sovik O, Kilimann MW. Mutations in the testis/liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nat Genet. 1996;14:337–40. [PubMed: 8896567]
- Maire I, Baussan C, Moatti N, Mathieu M, Lemonnier A. Biochemical diagnosis of hepatic glycogen storage diseases: 20 years French experience. Clin Biochem. 1991;24:169–78. [PubMed: 1645631]
- Morava E, Wortmann SB, van Essen HZ, Liebrand van Sambeek R, Wevers R, van Diggelen OP. Biochemical characteristics and increased tetraglucoside excretion in patients with phosphorylase kinase deficiency. J Inherit Metab Dis. 2005;28:703–6. [PubMed: 16151901]
- Ørngreen MC, Schelhaas HJ, Jeppesen TD, Akman HO, Wevers RA, Andersen ST, ter Laak HJ, van Diggelen OP, DiMauro S, Vissing J. Is muscle glycogenolysis impaired in X-linked phosphorylase b kinase deficiency? Neurology. 2008;70:1876–82. [PubMed: 18401027]
- Preisler N, Ørngreen MC, Echaniz-Laguna A, Laforet P, Lonsdorfer-Wolf E, Doutreleau S, Geny B, Akman HO, Dimauro S, Vissing J. Muscle phosphorylase kinase deficiency: a neutral metabolic variant or a disease? Neurology. 2012;78:265–8. [PubMed: 22238410]
- Qin L, Wang J, Tian X, Yu H, Truong C, Mitchell JJ, Wierenga KJ, Craigen WJ, Zhang VW, Wong LC. Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J Mol Diagn. 2016;18:446–53. [PubMed: 26944031]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rodríguez-Jiménez C, Santos-Simarro F, Campos-Barros Á, Camarena C, Lledín D, Vallespín E, Del Pozo Á, Mena R, Lapunzina P, Rodríguez-Nóvoa S. A new variant in PHKA2 is associated with glycogen storage disease type IXa. Mol Genet Metab Rep. 2017;10:52–5. [PMC free article: PMC5233919] [PubMed: 28116244]
- Roscher A, Patel J, Hewson S, Nagy L, Feigenbaum A, Kronick J, Raiman J, Schulze A, Siriwardena K, Mercimek-Mahmutoglu S. The natural history of glycogen storage disease types VI and IX: long-term outcome from the largest metabolic center in Canada. Mol Genet Metab. 2014;113:171–6. [PubMed: 25266922]
- Schneider A, Davidson JJ, Wullrich A, Kilimann MW. Phosphorylase kinase deficiency in I-strain mice is associated with a frameshift mutation in the alpha subunit muscle isoform. Nat Genet. 1993;5:381–5. [PubMed: 8298647]
- Tsilianidis LA, Fiske LM, Siegel S, Lumpkin C, Hoyt K, Wasserstein M, Weinstein DA. Aggressive therapy improves cirrhosis in glycogen storage disease type IX. Mol Genet Metab. 2013;109:179–82. [PMC free article: PMC3672367] [PubMed: 23578772]
- van Beurden EA, de Graaf M, Wendel U, Gitzelmann R, Berger R, van den Berg IE. Autosomal recessive liver phosphorylase kinase deficiency caused by a novel splice-site mutation in the gene encoding the liver gamma subunit (PHKG2). Biochem Biophys Res Commun. 1997;236:544–8. [PubMed: 9245685]
- van den Berg IE, van Beurden EA, de Klerk JB, van Diggelen OP, Malingré HE, Boer MM, Berger R. Autosomal recessive phosphorylase kinase deficiency in liver, caused by mutations in the gene encoding the beta subunit (PHKB). Am J Hum Genet. 1997;1997;61:539–46. [PMC free article: PMC1715950] [PubMed: 9326319]
- Wang J, Cui H, Lee NC, Hwu WL, Chien YH, Craigen WJ, Wong LJ, Zhang VW. Clinical application of massively parallel sequencing in the molecular diagnosis of glycogen storage diseases of genetically heterogeneous origin. Genet Med. 2013;15:106–14. [PubMed: 22899091]
- Wehner M, Clemens PR, Engel AG, Kilimann MW. Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Hum Mol Genet. 1994;3:1983–7. [PubMed: 7874115]
- Willems PJ, Gerver WJ, Berger R, Fernandes J. The natural history of liver glycogenosis due to phosphorylase kinase deficiency: a longitudinal study of 41 patients. Eur J Pediatr. 1990;149:268–71. [PubMed: 2303074]
- Wüllrich-Schmoll A, Kilimann MW. Structure of the human gene encoding the phosphorylase kinase beta subunit (PHKB). Eur J Biochem. 1996;238:374–80. [PubMed: 8681948]
- Wuyts W, Reyniers E, Ceuterick C, Storm K, de Barsy T, Martin JJ. Myopathy and phosphorylase kinase deficiency caused by a mutation in the PHKA1 gene. Am J Med Genet A. 2005;133A:82–4. [PubMed: 15637709]
- Zhang J, Yuan Y, Ma M, Liu Y, Zhang W, Yao F, Qiu Z. Clinical and genetic characteristics of 17 Chinese patients with glycogen storage disease type IXa. Gene. 2017;627:149–56. [PubMed: 28627441]
Publication Details
Author Information and Affiliations
University of North Carolina - Chapel Hill
Chapel Hill, North Carolina
Duke University Medical Center
Durham, North Carolina
Phildelphia, Pennsylvania
Department of Pediatrics
Duke University Medical Center
Durham, North Carolina
Duke University Medical Center
Durham, North Carolina
Publication History
Initial Posting: May 31, 2011; Last Update: November 1, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Herbert M, Goldstein JL, Rehder C, et al. Phosphorylase Kinase Deficiency. 2011 May 31 [Updated 2018 Nov 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.