Introduction
The research on liver-directed gene therapy and hepatocyte transplantation has progressed in parallel. Hepatocytes, with or without genetic modification have been used to introduce normal genes into patients or animal models with inherited disorders, while gene transfer can be used to expand hepatocytes in culture or abrogate allograft rejection. In this article, we discuss the current state of the two areas of research, as well as the hurdles that remain to be crossed for widespread application of these advances in the treatment of liver diseases.
Hepatocyte Transplantation
Although liver transplantation has dramatically improved the prognosis for patients with acute or end-stage liver failure, and inherited metabolic disorders, because of the complexity and associated morbidity and mortality of this procedure, hepatocyte transplantation is being explored as a convenient and safer alternative. Transplanting isolated hepatocytes by percutaneous or transjugular infusion into the portal vein, or injecting into the splenic pulp or the peritoneal cavity, is a less invasive procedure compared with liver transplantation. As the host liver is not removed or resected, the loss of graft function should not worsen liver function. Furthermore, isolated hepatocytes could be, potentially, cryopreserved for ready access.1 After extensive evaluation in experimental animals, clinical trials of hepatocyte transplantation have been initiated at several institutions for acute or chronic liver failure and inherited metabolic disorders. Hepatocytes transplantation has been explored as a vehicle for ex vivo gene therapy and is being considered for rescuing patients from radiation-induced liver damage resulting from radiotherapy for liver tumors.
Hepatocyte Transplantation for Liver-Based Metabolic Diseases
Hepatocyte transplantation has been tested on a number of animal models with liver-based metabolic disorders. The Gunn rat is an animal model of Crigler-Najjar syndrome type 1 (CN1). This mutant strain of Wistar rats lacks bilirubin-UDP-glucuronosyltransferase (UGT1A1) activity and consequently, accumulate toxic plasma levels of unconjugated bilirubin.2 Hepatocyte transplantation has been tested most extensively in Gunn rats and Nagase genetically analbuminemic (NAR) rats. In both models, transplantation of normal donor hepatocytes ameliorated the metabolic deficit.3 When the liver is structurally normal, which is the case in many inherited liver based disorders, hepatocytes injected into the splenic pulp or infused into the portal vein migrate to the liver and become integrated into the hepatic cords within days. These cells become morphologically identical to the host cells and function throughout life, and can be recognized only by virtue of biochemical or genetic markers.4,5 Hepatocyte transplantation has also resulted in partial correction of metabolic disorders in low density lipoprotein receptor-deficient6 Watanabe heritable hyperlipidemic rabbits (an animal model of familial hypercholesterolemia), and the Long-Evans Cinnamon rat (an animal model for Wilson's disease).7 Most functional liver proteins are present in great excess, therefore, the initial assumption was that the transplantation of a small fraction of the total hepatocyte mass (˜1–5%) should correct many metabolic disorders. However, most experimental and clinical studies have not borne out this expectation. Although repeated hepatocyte infusions could improve the response to transplantation to some extent,8 the most dramatic results have been obtained by massive preferential repopulation of the host liver by engrafted hepatocytes.
Massive Repopulation of the Liver
In inherited disorders which result in death of host hepatocytes, such as fumarylacetoacetate hydrolase (FAH) deficient mutant mice (a model of hereditary tyrosinemia type I) or transgenic mice overexpressing the plasminogen activator (uPA), transplanted normal hepatocytes can spontaneously repopulate the liver, eventually replacing almost all host hepatocytes.9 However, the transplant recipient mice remain susceptible to the development of hepatocellular carcinomas. Therefore, continuing cancer risk or recurrent disease should be considered in determining the clinical indications for hepatocyte transplantation for specific diseases.
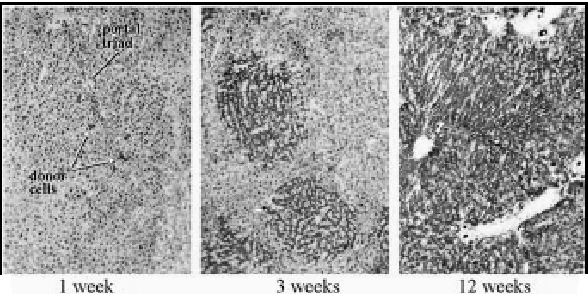
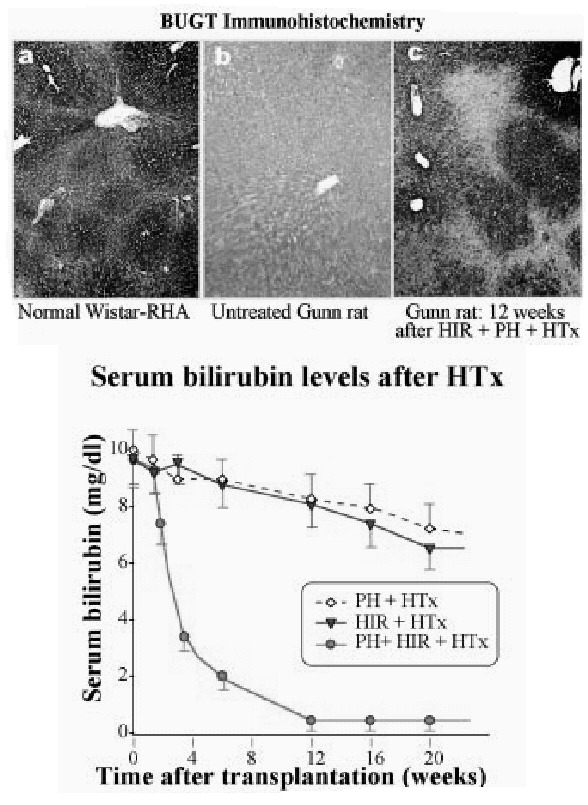
In most metabolic diseases, however, the host hepatocytes do not have a rapid turnover. In these cases, the host hepatocytes respond to proliferative stimuli to the same extent, as do the engrafted cells, precluding preferential growth of transplanted cells. Therefore, for a more general application, strategies are being developed to inhibit host hepatocyte regeneration, while providing a proliferative stimulus to the transplanted cells. In experimental model, the proliferative pressure is exerted by performing partial hepatectomy, causing reperfusion injury, inducing Fas-mediated apoptosis of host hepatocytes or administering pharmacological doses of thyroid hormone.10,11 For preventing the proliferation of host hepatocytes, a plant alkaloid, retrorsine, which blocks the hepatocyte cell cycle has been utilized.12 However, as retrorsine is potentially carcinogenic, X-irradiation of the liver has been evaluated as a part of the preparative regimen.13,14 In initial studies, the livers of recipient dipeptidylpeptidase IV-deficient rats were irradiated (50 Gy) and the rats were subjected to partial hepatectomy. Following transplantation of hepatocytes from congeneic normal rats (˜0.1% of the rat liver hepatocyte mass), the engrafted cells proliferated preferentially, forming small clusters in 3 weeks, and replacing nearly all host hepatocytes in 3 months (Fig. 1). Transplantation of normal hepatocytes from congeneic donor rats into Gunn rats that had undergone partial hepatectomy and preparative hepatic irradiation resulted in massive repopulation of the liver by the normal cells (Fig. 2), resulting in complete normalization of serum bilirubin levels.15 Interestingly, hepatocyte transplantation completely prevented radiation-induced morphological injury of the liver and the synthetic function of the liver was fully retained in the massively repopulated liver.14 Efforts to stimulate the proliferation of the transplanted hepatocytes by non-surgical means, such as controlled expression of FasL in the liver by gene transfer are underway.16


Clinical Experience with Hepatocyte Transplantation for Inherited Metabolic Disorders
Patients with CN1, ornithine transcarbamylase (OTC) deficiency and α1-antitrypsin deficiency have been transplanted with allogeneic hepatocytes. Two children with OTC deficiency showed significant evidence of the enzyme activity. However, one of these patients died shortly from hyperammonemia. In the other, hyperammonemia was corrected for a 10-day period, but recurred after that time, and liver transplantation was required for correction of the metabolic abnormality (I.J. Fox, personal communication). Direct evidence of function of transplanted hepatocytes was obtained in a CN1 patient (UGT1A1 deficiency).17,18 Allograft rejection was prevented with standard Tacrolimus and prednisone therapy. After transplantation, the serum bilirubin concentration declined to half of the pretransplantation level and UGT1A1 activity in a liver biopsy specimen increased to ˜5% of normal. Although the procedure did not cause portal hypertension or other significant complications, but the extent of replacement of hepatic UGT1A1 activity was not sufficient to eliminate the need for phototherapy.
Hepatocyte Transplantation for Acute Liver Failure
As the liver architecture usually remains normal in acute liver failure and the liver can, potentially, regenerate, hepatocyte transplantation provides a logical approach to provide temporary metabolic support to buy time for recovery of the liver. Hepatocyte transplantation dramatically improves the survival of rodents with acute liver failure induced by hepatotoxins, liver ischemia, or 90% hepatectomy.19,20 In pigs with ischemic acute liver failure, hepatocyte transplantation prevented the development of intracranial hypertension.21 However, hepatocyte transplantation in patients with acute liver failure has provided beneficial results only when used as a “bridge” while awaiting liver transplantation.
In initial clinical studies on patients with fulminant liver failure, human fetal hepatocytes were injected into the peritoneal cavity, resulting in improved survival in patients with grade III encephalopathy.22 However, since hepatocytes injected into the peritoneal cavity with a supporting scaffold have not been found to survive long-term, in subsequent studies, hepatocytes have been transplanted at other sites. Most trials have used adult hepatocytes. In one study, five out of seven transplant candidates who received hepatocyte infusion through the splenic artery survived to undergo subsequent liver transplantation.23 In clinical trials on patients who were not candidates for liver transplantation, hepatocytes infusion into the portal vein complications were not serious, but included pulmonary embolization with the hepatocytes, transient hemodynamic instability and sepsis.2426 In some recipients, engrafted hepatocytes were demonstrated within the spleens. Improvement in ammonia, prothrombin time, encephalopathy, cerebral perfusion pressure and cardiovascular stability has been reported anecdotally. Although these studies suggested that the hepatocyte transplantation had provided some benefit, unequivocal evidence of function of the engrafted cells has been difficult to obtain, probably because relatively small numbers of hepatocytes had been transplanted.
A part of the difficulty in translating the results of animal studies on acute liver failure to clinical application is that in contrast to the situation in animal models, liver regeneration is slower in patients with acute liver failure.27 The improvement in survival in experimental acute liver failure does not always imply engraftment, since injection of hepatocyte lysates or bone marrow cells also improve the animals. Recent studies on a mouse model of acute liver failure that more closely resembles the clinical situation suggest that repeated hepatocyte infusions may be required for improved patient survival.28
Hepatocyte Transplantation for Chronic Liver Failure
Animal Studies
Efficacy of hepatocyte transplantation has been evaluated mostly in animal models of hepatic encephalopathy induced by portacaval shunting. In rats with end-to-side portacaval shunts, intrasplenic transplantation of hepatocytes improved behavioral score and amino acid imbalance,29 and prevented ammonium chloride-induced hepatic coma.30 The engrafted hepatocytes proliferated in the spleen, forming structures resembling hepatic chords.
Hepatocyte transplantation has also been shown to improve metabolic abnormalities and to prolong survival in rats with decompensated stable liver cirrhosis, induced by chronic administration of phenobarbital and carbon tetrachloride.31 Hepatocyte transplantation into cirrhotic livers causes a severe and prolonged increase in portal pressure.32 Although some transplanted hepatocytes migrate into cirrhotic nodules after intraportal infusion, it unlikely that enough cells can engraft to significantly improve liver function. It is also uncertain whether the transplanted cells could function within the cirrhotic nodules. However, hepatocytes engrafted in the spleen have been shown to improve metabolic function in rats with stable end-stage cirrhosis.
Clinical Experience
In an early clinical trial on ten patients with liver cirrhosis, 10–600 million hepatocytes were harvested from the left lateral liver segments of the patients and were injected into the spleen, or infused into the splenic artery or the portal vein. In one patient, ascites and encephalopathy resolved and hepatocytes were detected in the spleen by 99mPMT-radioisotope uptake 11 months after transplantation.33 These studies suggested that hepatocytes within cirrhotic nodules could function when present in a different environment. In subsequent studies, up to ten billion allogeneic hepatocytes were infused into the splenic artery of patients with decompensated chronic liver disease. Nuclear scanning demonstrated the presence of engrafted hepatocytes in the spleen. The procedure was well tolerated and was associated with improvement in encephalopathy, hepatic protein synthesis and renal function.26 Unfortunately, the improvement of liver function following hepatocyte transplantation in chronic liver failure patients has been relatively poor, compared with that observed in animal models. The difference could have resulted from the fact that in animal experiments, hepatocytes were injected into the splenic pulps, whereas in most of the clinical studies the cells were infused into the splenic artery.26 Hepatocytes infused into arteries of rats are lost rapidly.34
Current Issues
Currently, less than 30% of the hepatocytes transplanted into the liver engraft. Since the number of hepatocytes that can be transplanted at one time is limited, better elucidation of the mechanism of hepatocyte engraftment is needed to augment the survival of the transplanted cells, thereby reducing the need for repeated cell transplantation. As most of the high quality donor livers are utilized for whole organ transplantation, it has been difficult to obtain well preserved livers for harvesting hepatocytes. Cryopreservation of high quality hepatocytes could provide ready access to the cells and could permit repeated hepatocyte transplantation from a single donor. At this time, however, cryopreserved hepatocytes have not been shown to engraft as well as fresh hepatocytes.35
Current technology does not permit long-term expansion of primary hepatocytes in vitro. Only the hepatocytes that have been immortalized by gene transfer are capable of long-term growth and correcting metabolic abnormalities in liver failure after transplantation.30,36 Xenogeneic cells could offer a soluion to the scarcity of human donor hepatocyts, but such cells face immunological processes that are different from that following allotransplantation.37,38 So far, the short survival of xenografts has precluded the determination as to whether organs or tissues from another species will functionally substitute for human organs.39 Transmission of infectious agents through xenogeneic liver cells also remains a potential concern.40 On the other hand, it is possible, that engrafted xenogeneic hepatocytes could be resistant to recurrence of infection by human-specific viruses.
Management of immunosuppression remains a special problem for hepatocyte transplantation, because in many cases detection of early rejection can be difficult. In some cases, continued functioning of the engrafted hepatocytes can be determined by direct biochemical analysis, e.g., analysis of pigments excreted in bile in the case of CN1. In other situations, viability of the transplanted cells may be difficult to determine despite repeated liver biopsies, because of the non-uniformity of the distribution of the transplanted cells in the liver. New non-invasive approaches to the detection of the transplanted hepatocytes are needed.
Liver-Directed Gene Therapy
Potential Indications
Liver-directed gene therapy is being contemplated for both inherited disorders and acquired conditions, such as infectious and neoplastic diseases, cirrhosis of the liver and immune rejection of transplants. Missing gene products causing inherited diseases could be replaced by transferring genes expressing those proteins. In other cases, specific genes could be overexpressed for therapeutic purposes, such as the overexpression of metalloproteases for the treatment of cirrhosis. In some situations, such as CN1 or low-density lipoprotein receptor deficiency (familial hypercholesterolemia), the missing gene product must be expressed in the liver for appropriate metabolic effect. In other cases, the large size of liver and secretory characteristics of the liver could be exploited to generate “biodrugs” for export out of the hepatocytes. Such proteins include coagulation factors, hormones or vaccines. In some cases, proteins that are normally expressed at extrahepatic sites could be expressed in the liver for specific purposes. For example, the catalytic subunit of the apolipoprotein B mRNA editing enzyme (APOBEC-1), which is normally expressed in the intestinal epithelial cells, could be expressed ectopically in the liver to switch the hepatic apolipoprotein production from apo B100 to apo B48, thereby reducing the production of low density lipoproteins.41 Similarly, hepatic expression of PDX, a homeobox protein that is responsible for pancreatic differentiation, could result in the secretion of insulin from hepatocytes.42 In other cases, nucleic acids may be used to inhibit the expression of deleterious proteins, such as viral proteins or mutant a1-antitrypsin. This could be accomplished by transfecting synthetic antisense RNAs, ribozymes43 or inhibitory double-stranded RNAi.44,45 Genes expressing antisense RNAs, RNAi, ribozymes or dominant negative proteins46 could be transferred into the liver. Finally, technologies are being developed to correct mutations within the endogenous genes in intact organisms. This could be accomplished by targeted replacement of the defective gene,47 or by site-directed correction of a target genomic sequence.48 These newer strategies permit the repaired gene to remain under the control of the endogenous promoter, whereby physiological regulation is retained. A partial list of inherited and acquired disorders targeted for liver-directed gene therapy is provided in Table 1. Gene therapies for cancer and infectious diseases of the liver pose some special problems, and have been discussed separately.
Table 1
Some liver-based disorders that are current targets for gene therapy.
Gene Therapy for Cancer
Nucleic acid transfer approaches are being designed to kill the tumor cells or inhibiting their growth, reduce tumor blood supply, evoke anti-tumor immune response, or to augment the effect of chemotherapy and radiotherapy. P53, a sentinel gene of the cell cycle, has been transferred to induce apoptosis of tumor cells.49 The herpes simplex virus thymidine kinase (HSV-TK), which converts a prodrug, ganciclovir, to toxic ganciclovir phosphate,50 or cytosine deaminase and purine nucleoside phosphorylase (which converts fludarabine to a diffusible toxic metabolite)51 have been expressed as a “suicide gene” in an attempt to ablate tumors. Ganciclovir phosphate may diffuse to neighboring cells, killing them by “by-stander effect”, extending number of tumor cells killed. To reduce the toxic effect on normal cells, the suicide genes have been targeted to tumor cells by tagging the DNA to monoclonal antibody directed at cell surface proteins that are preferentially expressed in tumor cells, such as AF-20, a 180-kDa tumor-specific cell surface glycoprotein, expressed in hepatoma cell lines.52 Another strategy to increase the therapeutic ration is to use tumor-specific promoters (e.g., alpha-fetoprotein or carcinoembryonic antigen) to drive the transgene expression. A different approach uses E1B-mutant adenoviruses that are replicate preferentially in P53-deficient tumor cells,53 although P53 deficiency may not be always required for the replication of these mutant adenoviruses.
Since killing the tumor cells by topical expression of toxic gene products does not eliminate tumors at metastatic sites, efforts are being made to inhibit neovascularization, that is needed for both primary and metastatic tumor growth, by the expressing angiostatin or endostatin genes.54,55
Host immune response against tumor antigens is important in eliminating tumor cells that are often present as micrometastasis. Expression of tumor-specific “neoantigens” may evoke this necessary immune response, provided they are presented properly to the immune system. Tumor antigens, such as those from melanoma cells, have been used for DNA-based tumor vaccination. Genetic manipulation of antigen-presenting cells may augment the host immune response against tumor cells. Since cytokines, such as TGFβ or IL10, secreted by large tumors, suppress immune response56 “debulking” the tumor by surgery, radiotherapy, chemotherapy or gene therapy may enhance the immune response.
Currently, the greatest potential of gene therapy for cancer is an adjunct to chemotherapy or radiotherapy. Irradiation of tumors augments tumor cell transduction using recombinant viruses. Transgene expression could be driven by radiation inducible promoters.57 On the other hand, inhibition of the expression in the tumor cells of radioprotective proteins, such as ATM (“mutated in ataxia telangiectasia”), could increase radiosensitivity of the tumors.58
Gene Therapy for Infectious Diseases
Nucleic acid-based approaches are being explored both for prophylaxis and treatment of hepatic viral infections. DNA vaccination59 can be safer and cheaper by eliminating the possibility of vaccine contamination infectious agents from tissue culture. The injected DNA itself can enhance the immunoreactivity of the expressed antigen. Expression of the antigenic peptides directly in antigen presenting cells could augment the immunoresponse by improving presentation. Expression of specific cytokines can be used to promote the proliferation of antigen presenting cells, as well as the maturation of T cells to helper T cells. Gene transfer is also being used to express single chain antibodies or antibody fragments in hepatocytes to make them resistant to viral infections.60
Gene therapy can also be used to interfere with the viral life-cycle by introducing synthetic antisense RNAs, ribozymes, or DNA ribonucleases (see below). Ribozymes, antisense RNAs or dominant negative proteins61 can also be expressed within the target cells by gene transfer
Gene Therapy for Inherited Disorders
As many inherited disorders are caused by the abnormality of a single gene, the effect of gene therapy can be evaluated directly and precisely in these conditions. For this reason, rare single gene abnormalities continue to be important targets of liver-directed gene therapy. Table 1 contains a partial list of inherited disorders that are currently targets of gene therapy.
Methods of Gene Delivery to the Liver
Genes may be delivered to the liver by systemic administration, infusion into the portal vein, hepatic artery or bile duct, or direct injection into the liver. Alternatively, hepatocytes isolated from the liver, or immortalized liver cells can be transduced with a therapeutic gene and then transplanted into the liver as a part of an “ex vivo” gene therapy approach. Commonly used methods for delivering genes to the liver of intact organisms, such as those using replication-deficient recombinant viruses, or non-viral vehicles are briefly discussed below. Frequently used methods of gene transfer to the liver are listed in Table 2.
Vectors Based on Recombinant Viruses
The attainable infectious titer, ability of the virus to infect non-dividing cells, integration into the host genome, repeatability of administration and safety of the vector system are important considerations in selecting recombinant viruses for gene therapy for specific diseases.
Retrovirus-Based Vectors
Proviral DNA, complimentary for the RNA genome of retroviruses, is integrated into the host genome and are transmitted to the progeny of the transduced cells.62 Tumor retroviruses, such as, Moloney's murine leukemia virus (MoMuLV), have been used extensively for generating recombinant vectors. The viral structural genes are replaced by target transgenes, leaving packaging signal (ψ) intact.63 The viral proteins are provided in trans by “packaging cell lines”, generating a replication-deficient recombinant virus. The envelope protein determines the range of species infectable by the recombinant retrovirus. The host range can be expanded by engineering proteins from other viruses, such as the G-protein of the vesicular stomatitis virus (VSV) into the retroviral envelope. After the recombinant virus enters the cell, the RNA genome is reverse-transcribed into double-stranded DNA provirus, which is transported to the nucleus and is integrated into the host genome. For the tumor viruses, this process requires resolution of the nuclear envelope that occurs during cell division. Therefore, the tumor retroviruses are inefficient in infecting primary hepatocytes, which undergo mitosis infrequently. Lentiviruses, which are retroviruses of the immunodeficiency group, form preintegration complexes that can be translocated into non-dividing nuclei. Therefore, recombinant lentiviruses are being developed for liver-directed gene therapy. However, it has been suggested that recombinant lentiviruses may need the hepatocytes to be in cell cycle for efficient gene transfer in vivo.64
Table 2
Advantages and limitations of liver-directed gene therapy methods.
Recombinant Adenovirus
Adenovirus types 5 and 2, are large linear double-stranded DNA viruses that are commonly used for preparing gene transfer vectors. Recombinant adenoviruses can transfer genes into both dividing and quiescent cells with high efficiency. Recombinant adenoviruses localize preferentially to the liver after intravenous administration in rodents.65 However, clinical trials in human subjects indicate transduction of human liver cells may not occur with such high efficiency.66 It is unclear whether this difference is based on the density of the adenoviral receptor, CAR, on the cell surface of hepatocytes of different species.
Adenoviral vectors are generated by disruption of the early region-1 (E1) by the insertion of the transgene. The E1 region encodes transcription factors required for the expression of adenoviral genes, and its disruption markedly inhibits the expression of the viral proteins. The recombinant virus is generated in packaging cells that provide the E1 gene products in trans. To abolish the possibility of viral gene expression and to increase the space available for insertion of the transgene, all structural genes of the virus have been deleted from the vectors.67 These “gene-deleted” vectors require helper adenoviruses to provide the structural proteins.
Cellular and humoral immune responses to adenoviral proteins limit the clinical application of adenovectors. In humans, antibodies are memory cells often exist from previous infections by the adenoviruses, and naïve individuals readily develop humoral and cell-mediated immunity against the viral antigens after the initial injection of the recombinant adenovirus. Neutralizing antibodies block gene transfer following subsequent administrations of the vector. Adenovirus-specific cytotoxic lymphocytes attack the host cells that are infected by the recombinant virus, resulting in liver damage and rapid loss of the transgene after secondary gene transfer.68 The helper-dependent, adenovectors, in which all viral genes deleted, retain their immunogenicity because of the viral proteins provided in trans by the packaging cells.69 Although these vectors express transgenes for a longer duration than do the first generation adenoviral vectors, secondary or tertiary administration fails to transfer the transgenes.
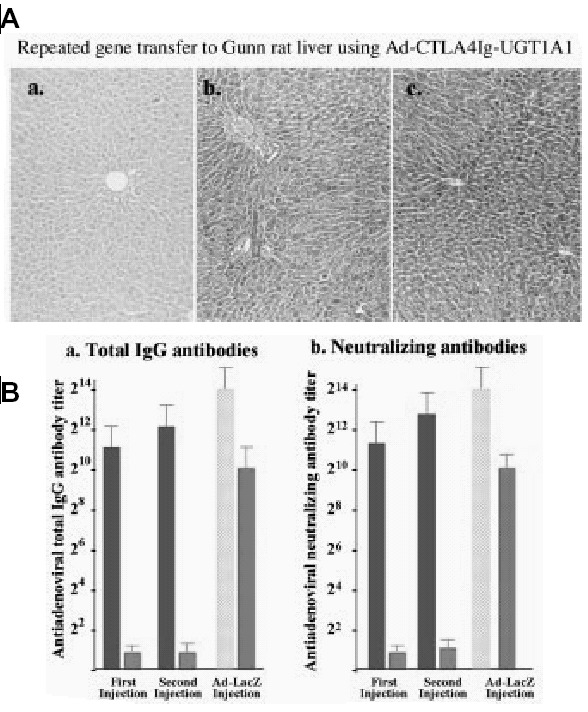
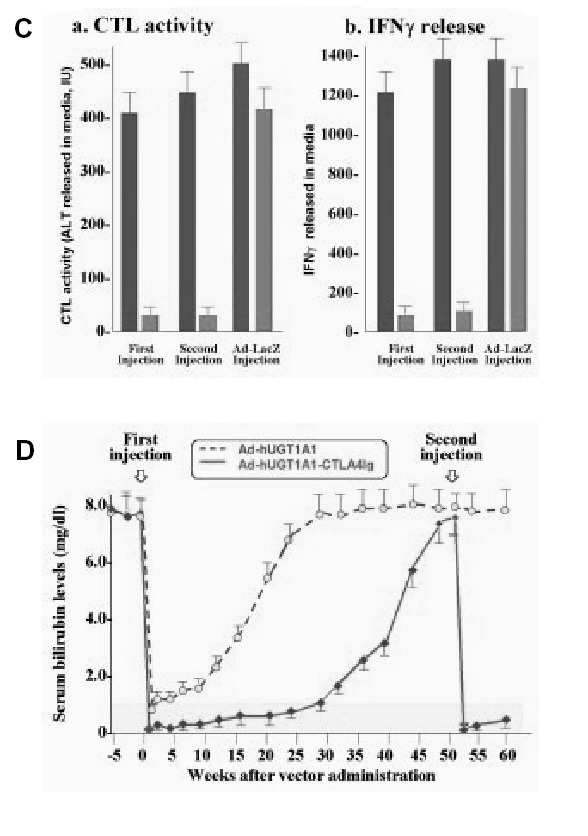
Development of immune response requires presentation of antigenic peptides by antigen presenting cells. Following docking of the antigen presenting cells with uncommitted T cells, the antigen presenting cells and the T cells costimulate each other via the B7-CD28 and CD40-CD40 ligand interactions. Inhibition of the B7-CD28 costimulation prevents effective immune response. CTLA4-Ig, a soluble inhibitory protein inhibits this costimulation (see Fig. 3). Injecting CTLA4-Ig alone at the time of administration of adenovectors does not prevent antibody formation.70 However, coexpression of CTLA4-Ig along with the target transgene permits multiple administration of the recombinant adenovirus.71 Host tolerance toward adenoviral proteins can be induced by injecting recombinant adenoviruses in utero or in newborn rats,65 inoculating adenoviral proteins into the thymus of young adult rats,72 or orally administering of small doses of adenoviral proteins.73 Since wildtype adenoviruses are human pathogens, safety concerns persist regarding tolerization of humans to adenoviral antigens remain. Adenoviral proteins may have additional toxic effects. A patient with ornithine transcarbamylase deficiency died shortly after recombinant adenovirus administration. The death was thought to have resulted from massive cytokine release, rather than an immune response to the virus.
As discussed above, the hepatotropism of recombinant adenoviruses that is seen in rodents was not found in human trials.74 Therefore, attempts are being made to modify the viral surface proteins to change their tropism, so that they can be internalized by hepatocytes through receptors that are normally present on the hepatocyte surface.75
Herpes Simplex Virus-1
Herpes simplex virus-1 (HSV-1) is a 150 kb double-stranded DNA virus with a broad host range76 that can infect both dividing and non-dividing cells. However, long-term gene expression in the liver has not been achieved with the currently available HSV vectors.
Recombinant Baculovirus
The Autographa californica nuclear polyhedrosis virus (AcNPV), which is usually used to generate recombinant proteins in insect cells,77 can infect hepatocytes, but not other mammalian cells and can express transgenes that are driven by mammalian or viral promoters.
Recombinant Adeno-associated Virus
The wildtype adeno-associated virus-2 (AAV-2) is a small (4.7 kb) single-stranded DNA parvovirus that integrates preferentially on the q13.4-ter arm of human chromosome 19.78 The AAV can remain latent for long periods, but can cause lytic infection following infection with a “helper virus,” such as adeno- or herpes simplex virus, or when the cell is exposed to genotoxic stimuli, e.g., UV light or irradiation.79 AAV type 2 is internalized following binding to a cell surface heparin sulfate proteoglycan. The 145-bp inverted terminal repeats (ITR) that flank the AAV genome are needed for integration into the host genome. Following internalization, AAV vectors may remain as episomes or may integrate into the host genome over several weeks. The viral Rep protein directs the site specificity of AAV integration into chromosome 19. Recombinant vectors that lack this gene, do not exhibit the site-specificity of integration and in fact may remain in the nucleus as episomes for prolonged periods.80
Infusion of high doses of the recombinant virus into the portal vein of factor IX-deficient dogs resulted in the appearance of 5% of normal levels of factor IX activity in plasma.81 Efficiency of AAV-mediated gene transfer to tumors could be augmented when used in combination with tumor irradiation or chemotherapy.82 Recombinant AAV causes a humoral immune response, which may be a problem if the vector needs to be readministered.
Simian Virus 40—based Vectors
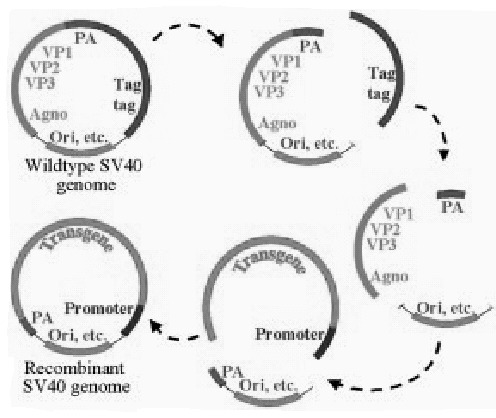
Simian virus 40 (SV40) is a non-enveloped 5.2 kb double-stranded DNA papova virus. The large (Tag) and small (tag) T antigens are transcription factors required for the expression of the viral structural genes, VP1, VP2 and VP3. The recombinant vector is generated by replacing the Tag genes of the viral genome by the target transgene. All three structural genes can be deleted to make additional space for inserting transgenes. The recombinant virus is generated by transfecting the recombinant viral genome into a helper cells (e.g., COS cells) that provide Tag in trans.83 Absence of the Tag gene makes the recombinant virus replication deficient and markedly reduces its immunogenicity. Up to 4.7 kb of exogenous DNA. can be insertd into SV40 vectors. The recombinant vector can be generated at 109 infectious units (IU)/ml and concentrated to 1012 IU/ml.84,85 Recombinant SV40 integrates into the genome of both dividing and non-dividing cells (see Fig. 4). The range of target cells is broad and includes hepatocytes, neural cells and bone marrow cells.8385

Non-Viral Vectors
Naked DNA
Injection of naked DNA into the muscle results in gene expression for several weeks. However, direct injection into the liver results in local inflammatory response and a shorter duration of expression. For experiments in mice or rats, the plasmid solution can be injected rapidly in a large volume, causing transient hepatic vascular congestion, which increases the transfection efficiency.86 As the injection of simple plasmids results in only short-term gene expression, this approach has little potential for clinical use. Modification of the plasmid for enhancing integration using a transposon-based mechanism has been discussed later.
Non-Viral Vectors
Non-viral vectors can be composed of lipid microcapsules (liposomes) or lipid-nucleic acid complexes (“lipoplex”), polycations, e.g., poly-L-lysine (PLL), polyethylenimine (PEI), polyglucosamines, lipopolyamines or cationic peptides, or polycation-lipid hybrids (“Lipopolyplex”). Lipopolyplexes are formed by compacting DNA constructs by addition of polycations, which may be then encapsulated into liposomes, forming particles that are smaller than liposomes and are better protected from nuclease degradation. Cationic lipid transfecting agents (e.g., lipofectamine, DOTAP), and cationic polymers, (e.g., poly-L-lysine and PEI) are used commonly for in vitro gene transfer,87 but are toxic in vivo or inactivated by plasma. For targeted gene delivery to hepatocytes, transfer vehicles have been modified to include ligands for the hepatocyte-preferred asialoglycoprotein receptor (ASGPr). Galactose-terminated peptides (e.g., Asialoorosomucoid or asialofetuin) or galactose can be conjugated to polylysine or PEI for targeted delivery to the liver.88 Liposomes composed of galactocerebrosides have also been used for receptor-mediated nucleic acid delivery to the liver.89
Inhibition of Gene Expression: Ribozymes, Antisense RNA, RANi and DNA Ribonucleases
Ribozymes are RNA enzymes that hybridize to complementary RNA sequences and catalyze endoribonucleolytic cleavage, causing rapid degradation of the RNA molecule.90 For hairpin ribozymes, the presence of a guanosine residue immediately downstream to the cleavage site is essential, but the effect is enhanced by a GUC sequence. Hammerhead ribozymes require only a UA, UC or UU dinucleotide for cleavage. Ribozymes can be synthesized and packaged for cellular uptake, or expressed in cells from transfected DNA. Recently, double stranded small ribonucleic acid molecules have been used effectively to degrade mRNA molecules and dramatically down-regulated the expression of specific genes. These inhibitory RNA molecules can be synthesized in vitro or expressed as hairpin molecules by transferring DNAs.
Hammerhead90 and hairpin91 ribozymes have been used to inhibit the production of hepatitis B and hepatitis C viruses in cultured cells. Following transfection of HuH7 cells (a differentiated human hepatoma line) with antisense oligonucleotides using ASGPr-polylysine complexes, the cells became resistant to HCV-directed protein synthesis.91 Multiple sites within a viral RNA genome can be targeted simultaneously using a series of ribozymes expressed from a single vector, thereby inhibiting the development of drug-resistant mutants.92 Since Hepatitis B virus replicates via a pregenomic RNA intermediate, it could also be a target for ribozyme-based therapy.90
DNA ribonucleases are synthetic single-stranded DNAs consisting of a 15-nucleotide catalytic domain, flanked by two RNA-binding domains. DNA ribonucleases cleave RNA with even more efficiency than do ribozymes.93 DNA ribonucleases have been reported to specifically cleave the hepatitis C viral genome93,94 and the RNA intermediates of hepatitis B.95
Homologous Recombination
In theory, site-specific recombination of DNA could be used to repair mutated or damaged DNA. Several proteins, including homologs of yeast recombination proteins rad51 and/or rad52, are important in homologous recombination in higher eukaryotes.96 Homologous recombination, which has been used extensively in cultured cells, has been rarely successful in vivo.96 Since homologous recombination is cell cycle-regulated, it occurs rarely in quiescent cells, such as hepatocytes.97
Triplex DNA
This method is based on generating a triple-chain DNA by the binding of a single stranded nucleic acid to the major groove of a homopurine region of the double stranded chromosomal DNA.98 For efficient triplex formation, the polypurine regions needs to 12–14 nucleotide long and guanine-rich. Cross-linking agents, such as psoralen or other mutagens can be attached covalently to the oligonucleotide.98 After intercalation of the psoralen at the target5' ApT3' site, UV-irradiation causes cross-linking of the thymines in the two strands. This substrate is then repaired by endogenous cellular DNA repair mechanisms, producing the characteristic T:A to A:T transversions. Single strand DNAs with cross-linking agents may also promote insertions and deletions via the excision repair pathway.99 Bifunctional oligonucleotides containing triple-helix-forming sequences, as well as conventional Watson and Crick base pairs have been used in culltured cells to cover a broader range of genomic sequences than is possible with conventional triple-helix forming oligonucleotides.100
Single Nucleotide Modification
This method utilizes the cellular mismatch repair machinery to correct genomic mutations. A synthetic oligonucleotide, complimentary to the targeted genomic DNA, except for a single mismatched base is synthesized. Most successful studies have utilized a DNA/RNA chimera. The RNA component, consisting of 2'-O-methyl ribonucleic acid residues, is included to increase the strength of hybridization with the targeted genomic DNA sequence. The mismatch between the oligonucleotide and the genomic DNA triggers the mismatch repair enzymes of the cell, permanently correcting the mutation. Plasmid mutation studies in E. coli strains deficient in specific repair proteins,101 indicated that this process required both RecA (an enzyme needed for homologous recombination) and MutS (a mismatch repair protein). Recombination and mismatch repair pathways are evolutionarily conserved and HuH-7 cell extracts, containing the mismatch repair protein hMSH2 can complement MutS-deficient E. coli in converting the mutant aminoglycoside resistance gene. Wild-type P53 may inhibit the initial pairing step in this repair process by inhibiting RecA and its human homolog Rad51.102
In intact rats, RNA-DNA chimeric oligonucleotides were delivered to the liver by receptor-mediated endocytosis, using lactosylated PEI or glactocerebroside-containing liposomes. The method has been shown to result in a Ser365Arg conversion of the rat factor IX gene103 and insertion of a guanosine base, which is deleted in the bilirubin-UDP-glucuronosyltransferase (UGT1A1) gene in jaundiced Gunn rats.104 In the latter case, expression of the wildtype functional enzyme was demonstrated and serum bilirubin levels were reduced significantly.104 Tagalakis and associates have demonstrated a high level of conversion of the gene expressing human apolipoprotein E2 in a transgenic mouse. The RNA-DNA chimera has also been used successfully for site-directed gene conversion of the apolipoprotein A2 gene in human lymphocytes,105 the nonsense mutation of carbonic anhydrase II in nude mouse primary kidney tubular cells106 and the missense mutation in the tyrosinase gene in albino mouse melanocytes.107
Transposon-Based Gene Delivery
Transposons are ubiquitous elements in eukaryotic genomes that can move from one location to another by a “cut-and-paste” mechanism. Emmons and associates identified transposable elements in fish genomes that had homology with the Tc1 transposon of the nematode Caenorhabditis elegans.108 These transposons of the Tc1/mariner superfamily were found to have short inverted repeats, flanking the coding sequences of a transposase. Hackett and associates generated a transposable element from an ancestral Tc1-like fish transposon that had acquired multiple mutations during evolution. This transposon was termed “Sleeping Beauty”, because it had remained dormant for millions of years and was made functional by correcting mutations in vitro. It is a 1.6-kb element, flanked by 250-bp terminal inverted repeats. DNA sequences, flanked by the terminal inverted repeats can efficiently integrate into mammalian cellular genome, in the presence of the transposase protein. The integration occurs at TA dinucleotide sites, which are duplicated upon insertion of the transposable element.109 The Sleeping Beauty transposon system has been used to effect integration of the coding region of human factor IX into the chromosomes of factor IX-deficient hemophilic mouse hepatocytes.110
Acknowledgments
This work was upported in part by NIH grants: RO1-DK 46057 (to JRC), RO1-DK 39137 (to NRC), the Liver Research Core Center grant DK-P30-41296 (Director, David A. Shafritz), The Gene Therapy Core of the Institute of Human Genetics of Albert Einstein College of Medicine and an American Cancer Society grant, RPG-00-066-01-CCE (to CG). The authors acknowledge the important contributions by many investigators in the fields of hepatocyte transplantation and liver-directed gene therapy that were not cited in this limited review.
References
- 1.
- Moshage HJ, Rijntjes PJ, Hafkenscheid JC. et al. Primary culture of cryopreserved adult human hepatocytes on homologous extracellular matrix and the influence of monocytic products on albumin synthesis. J Hepatol. 1988;7:34–44. [PubMed: 2460521]
- 2.
- Demetriou A, Levenson SM, Whiting J. et al. Replacement of hepatic functions in rats by transplantation of microcarrier-attached hepatocytes. Science. 1986;233:1190–1192. [PubMed: 2426782]
- 3.
- Demetriou A, Levenson SW, Whiting J. et al. Organization, morphology and function of microcarrier-attached transplanted hepatocytes in rats. Proc Natl Acad Sci USA. 1986;83:7475–7479. [PMC free article: PMC386741] [PubMed: 2429307]
- 4.
- Gupta S. et al. Permanent engraftment and function of hepatocytes delivered to the liver: Implications for gene therapy and liver repopulation. Hepatol. 1991;14:144–148. [PubMed: 2066062]
- 5.
- Gupta S, Rajvanshi P, Lee C-D. Integration of transplanted hepatocytes in host liver plates demonstrated with dipeptidylpeptidase IV deficient rats. Proc Natl Acad Sci (USA). 1995;92:5860–5864. [PMC free article: PMC41601] [PubMed: 7597042]
- 6.
- Eguchi S. et al. Treatment of hypercholesterolemia in the Watanabe rabbit using allogeneic hepatocellular transplantation under a regeneration stimulus. Transplantation. 1996;62:588–593. [PubMed: 8830820]
- 7.
- Irani A. et al. Correction of liver disease following transplantation of normal rat hepatocytes into Long-Evans Cinnamon rats modeling Wilson's disease. Mol Ther. 2001;3:302–309. [PubMed: 11273771]
- 8.
- Rozga J. et al. Repeated intraportal hepatocyte transplantation in analbuminemic rats. Cell Transplant. 1995;4:237–43. [PubMed: 7773557]
- 9.
- Overturf K, Al-Dhalimy M, Ou C. et al. Serial transplantation reveals the stem-cell-like regenerative potential of adult mouse hepatocytes. Am J Pathology. 1997;151:1273–1280. [PMC free article: PMC1858091] [PubMed: 9358753]
- 10.
- Ilan Y. et al. Massive repopulation of rat liver by transplantation of hepatocytes into specific lobes of the liver and ligation of portal vein branches to other lobes. Transplantation. 1997;64:8–13. [PubMed: 9233693]
- 11.
- Oren R. et al. Role of thyroid hormones in stimulating liver repopulation in the rat by transplanted hepatocytes. Hepatology. 1999;30:903–913. [PubMed: 10498641]
- 12.
- Laconi E. et al. Long-term, near-total liver replacement by transplantation of isolated hepatocytes in rats treated with retrorsine. Am J Pathol. 1998;153:319–29. [PMC free article: PMC1852941] [PubMed: 9665494]
- 13.
- Guha C. et al. Liver irradiation: a potential preparative regimen for hepatocyte transplantation. Int J Radiat Oncol Biol Phys. 2001;49:451–457. [PubMed: 11173140]
- 14.
- Guha C, Sharma A, Gupta S. et al. Amelioration of radiation induced liver damage in partially hepatectomized rats by hepatocyte transplantation. Cancer Research. 1999;59:5871–5874. [PubMed: 10606225]
- 15.
- Guha C, parashar B, Deb NJ. et al. normal hepatocyte correct serum bilirubin after repopulation of Gunn rat liver subjected to irradiation/partial resection. Hepatology. 2002;36:354–362. [PubMed: 12143043]
- 16.
- Takahashi M, Deb NJ, Kawashita Y. et al. A novel strategy for in vivo expansion of transplanted hepatocytes using preparative hepatic irradiation and FasL-induced hepatocellular apoptosis Gene Therapy 2002. In press.
- 17.
- Fox IJ. et al. Treatment of Crigler-Najjar syndrome type I with hepatocyte transplantation. New Eng J Med. 1998;338:1422–1426. [PubMed: 9580649]
- 18.
- Roy Chowdhury J, Strom SC, Roy Chowdhury N. et al. Hepatocyte transplantation in humans: Gene therapy and more. Pediatrics. 1998;102:647–648. [PubMed: 9738191]
- 19.
- Makowka L. et al. Reversal of toxic and anoxic induced hepatic failure by syngeneic, allogeneic, and xenogeneic hepatocyte transplantation. Surgery. 1980;88:244–53. [PubMed: 6994267]
- 20.
- Demetriou AA, Reisner A, Sanchez J. et al. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology. 1988;8:1006–1009. [PubMed: 3047034]
- 21.
- Arkadopoulos N. et al. Transplantation of hepatocytes for prevention of intracranial hypertension in pigs with ischemic liver failure. Cell Transplant. 1998;7:357–63. [PubMed: 9710304]
- 22.
- Habibullah CM, Syed IH, Qamar A. et al. Human fetal cell transplantation in patients with fulminant hepatic failure. Transplantation. 1994;58:951–952. [PubMed: 7940741]
- 23.
- Strom SC. et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation. 1997;63:559–69. [PubMed: 9047152]
- 24.
- Bilir B. et al. Hepatocyte transplantation in acute liver failure Liver Transpl 2000632–40.41-43 . [PubMed: 10648575]
- 25.
- Soriano H. et al. Hepatocellular transplantation (HCT) in children wiht fulminant liver failure (FLF). Hepatology. 1997;26:239A(443).
- 26.
- Strom S, Roy Chowdhury J, Fox I. Hepatocyte transplantation for the treatment of clinical disese. Seminars in Liver Disease. 1999;19:39–48. [PubMed: 10349682]
- 27.
- Miyazaki M. et al. Reversal of lethal, chemotherapeutically induced acute hepatic necrosis in rats by regenerating liver cytosol. Surgery. 1983;94:142–50. [PubMed: 6410523]
- 28.
- Braun K, Degen J, Sandgren E. Hepatocyte transplantation in a model of toxin-induced liver disease: variable therapeutic effect during replacement of damaged parenchyma by donor cells. Nat Med. 2000;6:320–326. [PubMed: 10700235]
- 29.
- Ribeiro J. et al. Intrasplenic hepatocellular transplantation corrects hepatic encephalopathy in portacaval-shunted rats. Hepatology. 1992;15:12–18. [PubMed: 1727787]
- 30.
- Schumacher IK. et al. Transplantation of conditionally immortalized hepatocytes to treat hepatic encephalopathy. Hepatology. 1996;24:337–43. [PubMed: 8690402]
- 31.
- Kobayashi N. et al. Hepatocyte transplantation in rats with decompensated cirrhosis. Hepatology. 2000;31:851–7. [PubMed: 10733539]
- 32.
- Gagandeep S. et al. Transplanted hepatocytes engraft, survive, and proliferate in the liver of rats with carbon tetrachloride-induced cirrhosis. J Pathol. 2000;191:78–85. [PubMed: 10767723]
- 33.
- Mito M, Kusano M. Hepatocyte transplantation in man. Cell Transplantation. 1993;2:65–74. [PubMed: 1466053]
- 34.
- Mito M, Kusano M, Ohnishi T. et al. Hepatocellular transplantation. Gastroenterol Jpn. 1978;13:480–90. [PubMed: 218863]
- 35.
- David P. et al. Engraftment and albumin production of intrasplenically transplanted rat hepatocytes (Sprague-Dawley), freshly isolated versus cryopreserved, into Nagase analbuminemic rats (NAR). Cell Transplant. 2001;10:67–80. [PubMed: 11294474]
- 36.
- Runge D, Michalopoulos G, Strom S. et al. Recent advances in human hepatocyte culture systems. Biochem Biophys Res Commun. 2000;274:1. [PubMed: 10903886]
- 37.
- Platt J. et al. Transplantation of discordant xenografts: a review of progress. Immunol Today. 1990;11:450–456. [PubMed: 2073317]
- 38.
- Ramirez P. et al. Life-supporting human complement regulatory decay accelerating factor transgenic pig liver xenograft maintains the metabolic function and coagulation in the nonhuman primate for up to 8 days. Transplantation. 2000;70:989–998. [PubMed: 11045632]
- 39.
- Kanazawa A, Platt J. Prospects for xenotransplantation of the liver. Semin Liver Dis. 2000;20:511–522. [PubMed: 11200419]
- 40.
- Platt J. New directions for organ transplantation. Nature. 1998;392:11–17. [PubMed: 9579856]
- 41.
- Greeve J, Jona VK, Roy-Chowdhury N. et al. Hepatic gene transfer of the catalytic subunit of the apolipoprotein B mRNA editing enzyme, APOBEC-1, leads to reduction of LDL in normal and Watanobe heritable hyperlipidemic rabbits. J Lipid Res. 1996;37:2001–2017. [PubMed: 8895066]
- 42.
- Ferber S, Halkin A, Cohen H. et al. panceatic and duodenalhomeobox gene 1 induces expression of insulin gene in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6:568–572. [PubMed: 10802714]
- 43.
- Ozaki I, Zern MA, Liu S. et al. Ribozyme-mediated specific gene replacement of the a1- antitrypsin gene in human hepatoma cells. J Hepatology. 1999;31:53–60. [PubMed: 10424283]
- 44.
- Novina CD, Murray MF, Dykxhoorn DM. et al. siRNA-directed inhibition of HIV-1 infection. Nature Medicine. 2002;8:681–686. [PubMed: 12042777]
- 45.
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. [PubMed: 11910072]
- 46.
- Scaglioni P, Malegari M, Takahashi M. et al. Use of dominant negative mutants of the hepadnaviral core protein as antiviral agents. Hepatology. 1996;24:1010–1017. [PubMed: 8903368]
- 47.
- Roy Chowdhury J. Prospects of liver cell transplantation and liver directed gene therapyIn: Seminars. In: Berk PD, ed.Liver DiseaseThieme:19991–6. [PubMed: 10349678]
- 48.
- Kmiec EB, Kren BT, Steer CJ. Targeted gene repair in mammalian cells using chimeric RNA/ DNA oligonucleotides In: Friedman T, ed.Development of Human Gene Therapy Cold Spring Harbork: Cold Spring Harbor Laboratory Press,1999643–670.
- 49.
- Roth JA, Nguyen D, Lawrence DD. et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nature Med. 1996;2:985–991. [PubMed: 8782455]
- 50.
- Kokoris MS, Sabo P, Adman ET. et al. Enhancement of tumor ablation by a selected HSV-1 thymidine kinase mutant. Gene Ther. 1999;6:1415–1426. [PubMed: 10467366]
- 51.
- Mohr L, Shankara S, Yoon S-K. et al. Gene therapy of hepatocellular carcinoma in vitro and in vivo in nude mice by adenoviral transfer of the Escherichia coli purine nucleoside phosphorylase gene. Hepatology. 2000;31:606–614. [PubMed: 10706550]
- 52.
- Mohr L, Schauer JI, Boutin RH. et al. Targeted gene transfer to hepatocellular carcinoma cells in vitro using a novel monoclonal antibody-based gene delivery. Hepatology. 1999;29:82–89. [PubMed: 9862854]
- 53.
- Harada JN, Berk AJ. p53-Independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J Virol. 1999;73:5333–5344. [PMC free article: PMC112589] [PubMed: 10364280]
- 54.
- Tanaka T, Cao Y, Folkman J. et al. Viral vector-targeted anti-angiogenic gene therapy utilizing an angiostatin complementary DNA. Cancer Res. 1998;58:3362–3369. [PubMed: 9699667]
- 55.
- Blezinger P, Wang J, Gondo M. et al. Systemic inhibition of tumor growth and tumor metastases by intramuscular administration of the endostatin gene. Nature Biotech. 1999;17:343–348. [PubMed: 10207881]
- 56.
- Fakhrai H, Dorigo O, Shawler DL. et al. Eradication of established intracranial rat gliomas by transforming growth factor b antisense gene therapy. Proc Natl Acad Sci USA. 1996;93:2909–2914. [PMC free article: PMC39733] [PubMed: 8610141]
- 57.
- Kawashita Y, Ohtsuru A, Kaneda Y. et al. Regression of hepatocellular carcinoma in vitro and in vivo by radiosensitizing suicide gene therapy under the inducible and spatial control of radiation. Human Gene Ther. 1999;10:1509–1519. [PubMed: 10395376]
- 58.
- Fan Z, Chakravarty P, Alfieri A. et al. Adenovirus mediated antisense ATM gene transfer sensitizes prostrate cancer cell to radiation. Cancer Gene Therapy. 2000;7:1307–1314. [PubMed: 11059687]
- 59.
- Encke J, zu Pulitz J, Geissler M. et al. Genetic immunization generates cellular and humoral immune responses against the nonstructural proteins of the hepatitis C virus in a murine model. J Immunol. 1998;161:4917–4923. [PubMed: 9794426]
- 60.
- zu Putlitz J, Skerra A, Wands JR. Intracellular expression of a cloned antibody fragment interferes with hepatitis B virus surface antigen secretion. Biochem Biophys Res Comm. 1999;255:785–791. [PubMed: 10049788]
- 61.
- Scaglioni P, Malegari M, Takahashi M. et al. Use of dominant negative mutants of the hepadnaviral core protein as antiviral agents. Hepatology. 1996;24:1010–1017. [PubMed: 8903368]
- 62.
- Verma IM, Somia N. Gene therapy-promises, problems and prospects. Nature. 1997;389:239–242. [PubMed: 9305836]
- 63.
- Kalpana GV. Retroviral vectors for liver-directed gene therapy. Semin Liver Dis. 1999;19:27–37. [PubMed: 10349681]
- 64.
- Park F, Ohashi K, Chiu W. et al. Efficient lentiviral transduction of liver requires cell cycling in vivo. Nature Genet. 2000;24:49–52. [PubMed: 10615126]
- 65.
- Takahashi M, Ilan Y, Sengupta K. et al. Induction of tolerance to recombinant adenoviruses by injection into newborn rats: Long term amelioration of hyperbilirubinemia in Gunn rats. J Biol Chem. 1996;271:26536–26542. [PubMed: 8900123]
- 66.
- Roy-Chowdhury J, Horwitz MS. Evolution of adenoviruses as gene therapy vectors. Mol Ther. 2002;5:340–344. [PubMed: 11945059]
- 67.
- Mitani K, Graham FL, Caskey CT. et al. Rescue, propagation, and partial purification of a helper virus-dependent adenovirus vector. Proc Natl Acad Sci USA. 1995;92:3854–3858. [PMC free article: PMC42060] [PubMed: 7731995]
- 68.
- Yang Y, Li Q, Ertl HCJ. et al. Cellular and humoral immune responses to viral antigen create barriers to lung-directed gene therapy with recombinant adenoviruses. J Virol. 1995;67:2004–2015. [PMC free article: PMC188865] [PubMed: 7884845]
- 69.
- Kafri T. et al. Cellular Immune response to adenoviral vector infected cells does not require de novo viral gene expression: implecation for gene therapy. Proc Natl Acad Sci USA. 1998;95:11377–11382. [PMC free article: PMC21650] [PubMed: 9736744]
- 70.
- Kay MA, Meuse L, Gown AM. et al. Transient immunomodulation with anti-CD40 ligand antibody and CTLA4Ig enhances persistence and secondary adenovirus-mediated gene transfer into mouse liver. Proc Natl Acad Sci USA. 1997;94:4686–4691. [PMC free article: PMC20785] [PubMed: 9114052]
- 71.
- Thummala NR, Ghosh SS, Lee SW. et al. A non-immunogenic adenoviral vector, coexpressing CTLA4Ig and bilirubin-uridinediphosphoglucuronateglucuronosyltransferase permits long-term, repeatable transgene expression in the Gunn rat model of Crigler-Najjar syndrome. Gene Therapy. 2002;9:981–990. [PubMed: 12101428]
- 72.
- Ilan Y, Attavar P, Takahashi M. et al. Induction of central tolerance by intrathymic inoculation of adenoviral antigens into the host thymus permits long term gene therapy in Gunn rats. J Clin Invest. 1996;98:2640–2647. [PMC free article: PMC507724] [PubMed: 8958229]
- 73.
- Ilan Y, Prakash R, Davidson A. et al. Oral tolerization to adenoviral antigens permits long term gene expression using recombinant adenoviral vectors. J Clin Invest. 1997;99:1098–1106. [PMC free article: PMC507919] [PubMed: 9062369]
- 74.
- Raper SE. et al. A pilot study of in vivo liver-directed gene transfer with an adenoviral vector in partial ornithinetranscarbamylase deficiency. Hum Gene Ther. 2002;13:163–175. [PubMed: 11779420]
- 75.
- Einfeld DA. et al. Reducing the native tropism of adenovirus vectors requires removal of both CAR and integrin interaction. J Virol. 2000;75:11284–11291. [PMC free article: PMC114713] [PubMed: 11689608]
- 76.
- Fung Y, Federoff HG, Brownlee M. et al. Rapid and efficient gene transfer in human hepatocytes by herpes viral vectors. Hepatology. 1995;22:723–729. [PubMed: 7657275]
- 77.
- Hoffmann C, Sandig V, Jennings G. Efficient gene transfer into human hepatocytes by baculovirus vectors. Proc Natl Acad Sci USA. 1995;92:10099–10103. [PMC free article: PMC40743] [PubMed: 7479733]
- 78.
- Samulski RJ, Zhu X, Xiao X. et al. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J. 1991;10:3941–3950. [PMC free article: PMC453134] [PubMed: 1657596]
- 79.
- Walz C, Schlehofer JR, Flentje M. et al. Adeno-associated virus sensitizes HeLa cell tumors to gamma rays. J Virol. 1992;66:5651–5657. [PMC free article: PMC289132] [PubMed: 1323717]
- 80.
- Song S, Laipis PJ, Berns KI. et al. Effect of DNA-dependent protein kinase on the molecular fate of the rAAV2 genome in skeletal muscle. Proc Natl Acad Sci USA. 2001;98:4084–4088. [PMC free article: PMC31183] [PubMed: 11274433]
- 81.
- Wang L, Nichols TC, Read MS. et al. Sustained expression of therapeutic level of factor IX in hemophilia B dogs by AAV-mediated gene therapy in liver. Mol Ther. 2000;1:154–158. [PubMed: 10933925]
- 82.
- Alexander IE, Russell DW, Miller AD. DNA-damaging agents greatly increase the transduction of nondividing cells by adeno-associated virus vectors. J Virol. 1994;68:8282–8287. [PMC free article: PMC237296] [PubMed: 7966621]
- 83.
- Strayer DS, Zern MA, Roy Chowdhury J. What can SV40-derived vectors do for gene therapy? Curr Opin Mol Ther. 2002;4:313–323. [PubMed: 12222869]
- 84.
- Sauter BV, Parashar B, Roy Chowdhury N. et al. A replication deficient rSV40 mediates liver-directed gene transfer and a long-term amelioration of jaundice in Gunn rats. Gastroenterology. 2000;119:1348–1357. [PubMed: 11054394]
- 85.
- Strayer DS, Branco F, Zern MA. et al. Durability of Transgene Expression and Vector Integration: Recombinant SV40-Derived Gene Therapy Vectors Mol Ther 2002. In press. [PubMed: 12161189]
- 86.
- Liu F, Song YK, Liu D. Hydrodynamics-based transfectionin animals by systemic administrationof plasmid DNA. Gene Ther. 1999:61258–1266. [PubMed: 10455434]
- 87.
- Zabner J, Fasbender AJ, Moninger T. et al. Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem. 1995;270:18997–19007. [PubMed: 7642560]
- 88.
- Findeis MA, Wu CH, Wu GY. Ligand-based carrier systems for delivery of DNA to hepatocytes. 1994. pp. 341–351. [PubMed: 7898362]
- 89.
- Kren BT, Bandyopadhyay P, Roy Chowdhury N. et al. Oligonucleotide-mediated site-directed gene repair. Meths Enzymol. 2002;346:14–35. [PubMed: 11883065]
- 90.
- Sakamoto N, Wu CH, Wu GY. Intracellular cleavage of hepatitis C virus RNA and inhibition of viral protein translation by hammerhead ribozymes. J Clin Invest. 1996;98:2720–2728. [PMC free article: PMC507736] [PubMed: 8981917]
- 91.
- Welch PJ, Tritz R, Yei S. et al. Intracellular application of hairpin ribozyme genes against hepatitis B virus. Gene Ther. 1997;4:736–743. [PubMed: 9282175]
- 92.
- Welch PJ, Yei S, Barber JR. Ribozyme gene therapy for hepatitis C virus infection. Clin Diag Virol. 1998;10:163–171. [PubMed: 9741642]
- 93.
- Asahina Y, Ito Y, Wu CH. et al. DNA ribonucleases that are active against intracellular hepatitis B viral RNA targets. Hepatology. 1998;28:547–554. [PubMed: 9696023]
- 94.
- Wu CH, Wu GY. Targeted inhibition of hepatitis C virus-directed gene expression in human hepatoma cell lines. Gastroenterology. 1998;114:1304–1312. [PubMed: 9609768]
- 95.
- Pan WH, Devlin HF, Kelly C. et al. A selection system for identifying accessible sites in target RNAs. Rna-A Publication of the Rna Society. 2001;7(4):610–621. [PMC free article: PMC1370114] [PubMed: 11345439]
- 96.
- Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1997;27:1223–1242. [PMC free article: PMC148307] [PubMed: 9973609]
- 97.
- Yamamoto A, Taki T, Yagi H. et al. Cell cycle-dependent expression of the mouse Rad51 gene in proliferating cells. Mol Gen Genet. 1996;251:1–12. [PubMed: 8628240]
- 98.
- Chan PP, Glazer PM. Triplex DNA: fundamentals, advances, and potential applications for gene therapy. J Mol Med. 1997;75:267–282. [PubMed: 9151213]
- 99.
- Faruqi AF, Datta HJ, Carroll D. et al. Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol Cell Biol. 2000;20:990–1000. [PMC free article: PMC85216] [PubMed: 10629056]
- 100.
- Culver KW, Hsieh W-T, Huyen Y. et al. Correction of chromosomal point mutations in human cells with bifunctional oligonucleotides. Nature Biotechnol. 1999;17:989–993. [PubMed: 10504700]
- 101.
- Kren BT, Metz R, Kumar R. et al. Gene repair using RNA/DNA oligonucleotides. Sem Liver Dis. 1999;19:93–104. [PubMed: 10349687]
- 102.
- Sturzbecher H-W, Donzelmann B, Henning W. et al. p53 is linked directly to homologous recombination processes via RAD51/RecA protein interaction. EMBO J. 1996;15:1992–2002. [PMC free article: PMC450118] [PubMed: 8617246]
- 103.
- Kren BT, Bandyopadhyay P, Steer CJ. In vivo site-directed mutagenesis of the factor IX gene by chimeric RNA/DNA oligonucleotides. Nature Med. 1998;4:285–290. [PubMed: 9500600]
- 104.
- Kren BT, Parashar B, Bandyopadhyay P. et al. Correction of the UDP-glucuronosyl-transferase gene defect in the Gunn rat model of Crigler-Najjar syndrome type I with a chimeric oligonucleotide. Proc Natl Acad Sci USA. 1999;96:10349–10354. [PMC free article: PMC17891] [PubMed: 10468611]
- 105.
- Tagalakis AD, Graham IR, Riddell DR. et al. Gene correction of the apolipoprotein (Apo) E2 phenotype to wild-type ApoE3 by in situ chimeroplasty. J Biol Chem. 2001;276:13226–13230. [PubMed: 11278248]
- 106.
- Lai L-W, Chau B, Lien Y-H. In vivo Gene targeting in carbonic anhydrous II deficient mice by chimeric RNA/DNA oligonucleotides Conference Proceedings: 2nd Annual Meeting of the American Society of Gene TherapyWashington DC:1999236a101.
- 107.
- Alexeev V, Igoucheva O, Domashenko A. et al. Localized in vivo genotypic and phenotypic correction of the albino mutation in skin by RNA-DNA oligonucleotide. Nature Biotechnol. 2000;18:43–47. [PubMed: 10625389]
- 108.
- Radice AD, Bugaj B, Fitch DHA. et al. Wdespread occurance of TC1 transposon family: TC1-like transposons from teleost fish. Mol Gen Genet. 1994;244:606–612. [PubMed: 7969029]
- 109.
- Ivics Z, Hackett PB, Plasterk RH. et al. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. [PubMed: 9390559]
- 110.
- Yant SR, Meuse L, Chiu W. et al. Somatic integration and long-term transgene expression in normal and hemophilic mice using a DNA transposon system. Nature Genetics. 2000;25:35–41. [PubMed: 10802653]
Publication Details
Author Information and Affiliations
Authors
Chandan Guha, Siddhartha S. Ghosh, Sung W. Lee, Namita Roy Chowdhury, and Jayanta Roy Chowdhury.Copyright
Publisher
Landes Bioscience, Austin (TX)
NLM Citation
Guha C, Ghosh SS, Lee SW, et al. Hepatocyte Transplantation and Liver-Directed Gene Therapy. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.