NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.
The arena of Parkinson's disease (PD) genetics is becoming more and more crowded with findings emerging from complementary approaches. Classical linkage analyses in families with Mendelian inheritance, affected-pairs analyses in smaller families, and studies in genetically isolated populations are all contributing different pieces of what is emerging as a complex etiopathogenetic mosaic.
Here, we first focus on the novel monogenic forms (PARK6 to PARK9), which have been recently identified, and on DJ-1, the gene defective at the PARK7 locus. We then review the genome screens performed in the common, late-onset forms of the disease. These screens have delineated novel loci on chromosome 1p (PARK10) and 2q, and a list of other candidate regions for susceptibility or modifier genes. Further efforts are now needed to replicate/expand these findings and delineate/refine the regions of genuine linkage, in order to identify the biologically relevant genetic variants associated with, or modifying the PD phenotype.
Introduction
Despite PD being the second most common neurodegenerative disorder,1 the molecular genetic approaches have taken off with delay in comparison with other diseases, perhaps because PD had long been considered nongenetic in essence. However, this gap is rapidly being filled. The current catalogue of monogenic PD already includes ten entries (Table 1) with additional forms likely to be identified soon. Although they explain a very small fraction of PD cases at population level, monogenic forms are extremely useful in delineating molecular pathways that might be involved in the common forms of PD as well.2,3 Moreover, a considerable amount of data is being generated in genome-wide screens addressing directly the problem of the genetic bases of the common, nonMendelian form(s) of the disease.
In the current scenario, the short arm of chromosome 1 is emerging as a hot spot for PD-related genes (fig. 1), as three novel loci for autosomal recessive forms of early onset (AREP) (PARK6, PARK7, and PARK9), and a locus controlling susceptibility to, and/or onset age of late-onset PD (PARK10) have been mapped to chromosome 1p by five different groups.4-8 Recently, the defective gene at the PARK7 locus, DJ-1, has been identified.9
The PARK7 Locus and the DJ-1 Gene
In the framework of a large research project for gene mapping in a genetically isolated population of South West of The Netherlands (Genetic Research in Isolated Population, GRIP project), a large consanguineous pedigree was ascertained with four members affected by early-onset parkinsonism (the onset varying from age 27 to 40). A genome-wide scan in this family using homozygosity mapping yielded significant evidence for linkage to the chromosome 1p36 region, and this locus was named PARK7.5 Linkage to the very same region was later confirmed in a consanguineous pedigree from Central Italy with three affected members.10 Fine mapping studies and a positional cloning strategy led us to the identification of mutations in the DJ-1 gene in the two definitely linked families (fig. 2).9
All patients of the Dutch kindred carry a homozygous deletion, which removes ~14 kb of genomic sequence, including 4 kb upstream of the DJ-1 start codon, and a large part of the coding region of DJ-1. In the Italian family, a homozygous missense mutation replaces the highly conserved Leucine at position 166 with Proline (L166P) in the DJ-1 protein. Computer-assisted modeling suggested a globular, α/β-sandwich to be the most likely structure for DJ-1, with the L166 lying in the carboxy-terminal α-helix (fig. 3). The L166P replacement likely destabilizes this helix in the mutant DJ-1. Transfection experiments with constructs carrying the wild type or the L166P mutant lent further support to the argument that the L166P mutation is pathogenic, because this mutation dramatically changed the subcellular localization of the protein.9 In both COS and PC12 cell lines the wild-type protein showed a diffuse cytoplasmic and nuclear localization, a pattern which was previously reported by other groups. In contrast, the protein carrying the L166P mutation still showed a diffuse nuclear localization, but it lost the diffuse cytosolic pattern, appearing mainly colocalized with the mitochondria.9 The homozygous deletion found in the patients of the Dutch family represents a natural knockout of DJ-1, clearly indicating that the loss of function of DJ-1 is pathogenic. Moreover, the fact that the L166P DJ-1 mutant protein is still present in the nucleus, suggests that the loss of some currently unknown cytosolic function of DJ-1 is most likely pathogenic.
Human DJ-1 extends over 24 kb on genomic level, and it is organized in eight exons (fig. 2). The first two exons (named 1A and 1B) are noncoding and alternatively spliced in the mRNA. Northern blot analysis showed that DJ-1 is ubiquitously and highly expressed in the brain areas and extra-cerebral tissues.9 Interestingly, using Northern analysis, the expression seems more abundant in subcortical regions of the brain than in cortical areas. However, further work is needed to explore the regional pattern of DJ-1 expression at the protein level.
The genomic structures of the mouse and human DJ-1 genes are similar, and human and mouse DJ-1 proteins display 90% amino acid identity.11 Analysis of promoter activity using deletion constructs and luciferase assays in HeLa cells identified an Sp1 (Specificity Protein1, a transcription factor) dependent site at position _100 from the transcription initiation site contributing most of the promoter activity.11
The human DJ-1 protein consists of 189 amino acids and belongs to the ThiJ/PfpI family. Several members of this family of proteins are known in prokaryotes: ThiJ, involved in the thiamine synthetic pathway, PfpI and other proteases, araC and other transcription factors, and the glutamine amidotransferase family, which includes some bacterial catalases.
The exact function of the DJ-1 protein is unknown. However, data from the literature provide clues for its possible functions. A human cDNA termed DJ-1 was first cloned in 1997, and the encoded protein was reported to have oncogenic properties in cooperation with the oncogene ras.12 Two years later a human protein named RS that was identical to DJ-1 was identified as the regulatory (inhibitory) subunit of an RNA-binding protein (RBP) complex.13 In these studies DJ-1 immunoreactivity was shown in the cytoplasm and the nucleus, depending on the cell-cycle phase, or the cell-types studied. Furthermore, two independent groups identified a rat protein called CAP1 (Contraception Associated Protein 1) or SP22 (Sperm Protein 22), putatively involved in spermatogenesis and fertilization, as the rat DJ-1 homolog.14,15 In a yeast two-hybrid system, PIASxa/ARIP3, a negative modulator of the androgen receptor (AR), was detected as a DJ-1 binding protein, suggesting DJ-1 to be a positive regulator of AR-mediated transcriptional activity.16 PIASxα/ARIP3 belongs to a family of proteins (PIAS, Protein Inhibitor of Activated STAT) that modulate the activity of transcription factors by functioning as SUMO-1 (Small Ubiquitin-like Modifier) ligases.17 Interestingly, a DJ-1 mutant at Lys130 (K130R) is unable to regulate the AR activity.16 As the Lysine 130 of DJ-1 seems to be sumoylated, it is possible that this modification is necessary for the full activity of DJ-1.
Recent studies showing that human DJ-1 is converted into a variant having more acidic pI in response to oxidative stress (including the herbicide Paraquat or hydrogen peroxide) suggested a role for DJ-1 as an antioxidant.18,19 Intriguingly, the yeast DJ-1 homologue YDR533C is transcribed during the oxidative stress response and during the response to stress induced by misfolded proteins (which in turn is associated with oxidative stress),20,21 raising the question of whether DJ-1 also plays a role as a molecular chaperone. On the basis of the available evidence, DJ-1 might be involved in the cellular response to stress at multiple levels.9 First, it might directly react to stress signals (e.g., redox changes, misfolded proteins) by a chemical shift and/or change in multimerization state. Second, DJ-1 might modulate the gene-expression of the stress-response at post-transcriptional level, by the known interaction with RNA-binding protein complexes. Interestingly, the post-transcriptional regulation of gene expression is important for both neuronal function and spermatogenesis. Third, DJ-1 might translocate to the nucleus in response to stress signals. In the nucleus, it might interact with PIAS or other cofactors, and modulate the gene expression at transcriptional level. Although an involvement of human DJ-1 in the oxidative stress response, or in the response to misfolded protein stress remains to be proven and the exact function of DJ-1 is still unknown, the proposed model is intriguing in the light of the evidence of oxidative stress and protein misfolding documented in the brains of patients with PD.22,23 Recent studies have shown that mutations in alpha-synuclein and parkin, two other PD-related genes, might also be linked to oxidative stress.24,25
Future mutational analyses in large series of families and isolated early onset PD will reveal the frequency of DJ-1 mutations and allow accurate genotype-phenotype correlation studies. An autosomal dominant form of focal-segmental dystonia maps to 1p36 (the DYT13 locus).26 The telomeric part of the DYT13 critical interval overlaps for 6 cM with the centromeric part of the PARK7 interval, and the DJ-1 gene is located in the region of overlap. So far, linkage to this region has not been replicated in other dystonia families, and parkinsonian features have not been detected in the original DYT13-linked family. However, dystonic features such as laterocollis and blepharospasm are present in both the DJ-1-related families. This suggests that despite the differences in transmission pattern and clinical phenotypes, PARK7 and DYT13 might be allelic disorders; a mutational screening of the DJ-1 gene is therefore warranted in DYT13-linked families.
Pathological analysis of brains from patients with DJ-1-related forms is of great importance, but brain material is not currently available. Moreover, investigating the presence of the DJ-1 protein in brain from patients with Lewy body disease, as well as other neurodegenerative diseases is urgently warranted. These studies might provide important clues on the involvement of DJ-1 in common forms of neurodegeneration. The effect of mutations in DJ-1, either alone or in combination with mutations in other PD-related genes, is now being investigated in cell culture and animal models, including mice and flies. In transgenic flies, it will be possible to rapidly screen for genetic modifiers, either enhancers or suppressors of any resulting DJ-1-related phenotype. Furthermore, biochemical studies have been initiated in order to understand further the function of DJ-1, identify its interacting partners, and explore possible relationships between DJ-1 and the proteins encoded by the other genes which are firmly implicated in PD: α-synuclein and parkin. This work will help clarifying the pathogenesis of DJ-1-related disease, but, more importantly, it can provide new insights for understanding of the molecular mechanisms of classical PD.
The PARK6 Locus
This locus was first identified in a large Italian consanguineous family originating from Sicily.4 Four family members developed early-onset PD with onset ages ranging from 32 to 48 years. A genome-wide scan provided significant evidence for linkage to the short arm of chromosome 1, in the region 1p35-p36. Haplotype analysis delineated a 12.5 cM homozygously inherited region in the affected members, flanked by the markers D1S483 telomerically and D1S247 centromerically. A subsequent study analysed 28 smaller European AREP families, which did not carry mutations in the parkin gene. Eight out of 28 families supported linkage to PARK6, suggesting this as a frequent locus.27 Of particular interest is a second consanguineous family from Central Italy, in which a recombination event would reduce the critical region to 9 cM. However, none of these families independently had enough statistical power to detect significant linkage. Considering the genetic heterogeneity of AREP and PD, the assignment of these families to PARK6 should therefore be considered as provisional.
Early onset of parkinsonism, absence of atypical signs, slow disease progression, and good response to levodopa are unifying clinical features in different AREP forms, including parkin-related, DJ-1 related, and PARK6-linked families.5,10,28-31 This will make their differentiation difficult on clinical grounds, indicating the importance of genetic testing. In comparison with classical PD, the phenotype in the Dutch and Italian DJ-1-related families is characterized by an early onset and a slow progression. Interestingly, behavioral and psychic disturbances, and dystonic features (including blepharospasm) have been reported in both families, and they might represent “red flags” for suspecting DJ-1 involvement.5,10,31 Moreover, the average age at onset is in the early thirties for the parkin- and DJ-1-related families, whereas the onset in PARK6-linked families might be slightly later (late thirties - early forties). The pathology of PARK6-linked and DJ-1-related forms is unknown. Recent positron emission tomography studies in PARK6-linked and DJ-1-related patients showed: more uniform patterns of caudate/putamen dopaminergic terminal dysfunction in comparison with the one observed in classical PD; greater dopaminergic dysfunction than the one expected on the basis of clinical severity; a mild dysfunction in asymptomatic heterozygous PARK6 carriers.28,31 All these features also resemble the pattern observed in parkin-related disease, suggesting that these three AREP forms might be more similar to each other than to classical PD on pathophysiological grounds.
The similarities between PARK6- and DJ-1-associated phenotypes, and the proximity of the two loci on chromosome 1p, raised the question of whether mutations in similar or functionally related genes underlie both forms. An alternative possibility is that, due to a large genomic rearrangement occurring in one of the linked families, linkage is found in seemingly different regions, while there is only one underlying, defective gene. To explore this possibility, a mutational analysis of the DJ-1 gene should also be performed in the original PARK6-linked family.
Kufor-Rakeb Syndrome and the PARK9 Locus
This form has been initially described in a consanguineous family from a small community (Kufor-Rakeb) in the North of Jordan.32 In this kindred, five siblings developed a multisystemic neurodegenerative disease resembling the so-called “pallido-pyramidal” syndromes, and clinically characterized by onset below age of 20, akinetic-rigid parkinsonism (without tremor), pyramidal tract signs, supranuclear ophtalmoparesis, and cognitive deterioration. The progression is reported to be rapid, but the parkinsonian symptoms responded very well to levodopa therapy. Brain MRI showed progressive atrophy, starting from the pallidum and pyramids but later generalized. No pathology studies have been reported.32 A genome-wide scan in this family detected significant evidence for linkage to the 1p36 region,6 and despite the fact that multisystemic Kufor-Rakeb syndrome appears far from PD, this locus has been designated as PARK9. Furthermore, the PARK9 critical interval overlaps with the DYT13 and PARK6 critical regions at the telomeric and the centromeric border, respectively (fig. 1). PARK9 can therefore be allelic to one of these disorders. Cloning of the responsible genes at these loci will provide the answer to this question.
The PARK8 Locus
This novel locus for autosomal dominant PD was identified in a large pedigree, named the “Sagamihara family” from the region of origin in Japan.33 Clinical features in affected individuals of the kindred are reported to resemble very closely classical PD, with an average disease onset at 51 ± 6 years.34 Yet, a pattern of “pure nigral degeneration” without Lewy bodies was found at autopsy in four PD patients examined. Moreover, one carrier of the disease-linked haplotype developed pathologically-confirmed striatonigral degeneration, an atypical form of parkinsonism which is known to be associated with α-synuclein-positive brain pathology.33
In this family, a genome-wide scan yielded significant evidence for linkage of PD to the centromeric region of chromosome 12. Haplotype analysis suggests an incomplete penetrance of the mutation. This is of interest because an incomplete, age-related penetrance could have masked a Mendelian pattern in other smaller PD families. Interestingly, linkage to the same region was found in a genome scan for Alzheimer's disease (AD), the locus being termed AD5, but this linkage was mostly supported by the subset of AD families containing at least one affected individual with Lewy body pathology (Lewy body dementia).35 Lastly, within a genome scan in a set of 22 families with parkin gene mutations, the centromeric region of chromosome 12 yielded the strongest evidence of linkage.36 Taken together, these findings suggest that a single locus in this region might control susceptibility to a wide range of clinical and pathological phenotypes. Confirmation of linkage to PARK8 in other PD families has not been reported yet, and the defective gene at this locus remains unknown.
Genome-Wide Screens in Classical PD
Families segregating PD as a monogenic trait, and large enough to be suitable for classical linkage mapping are very rare. Moreover, for the vast majority of cases parkinsonism runs in families without a clearcut Mendelian pattern and highly variable expression, suggesting a complex multifactorial etiology. In this scenario, it has been suggested that it might be more feasible to perform genome-wide scans in series of small PD families containing 2 affected relatives (sibling pairs or other relative pairs).37 Such families are not rare, and collecting large numbers is not difficult in the framework of multicentric consortia. In these studies, nonparametric, model-free methods are often used. These methods are very robust because they do not require a genetic model to be specified.37 The methods are based on the general principle of comparing the observed number of alleles shared identical-by-descent (IBD) between pairs of affected relatives versus the numbers expected under the null hypothesis of no linkage. For example, at a given locus, the distribution of IBD sharing by sibling pairs under the null hypothesis is: 0 alleles (in 25% of the pairs), 1 allele (50% pairs), and 2 alleles (25% pairs). Any deviation from this distribution in the direction of an excess of allele sharing is considered as evidence of linkage, and the results can be expressed as LOD scores as in classical linkage analysis. As for all available methods this approach is not free from limitations and caveats. Since a large number of families are used, genetic heterogeneity influences greatly the outcome of such studies. Depending on the number and magnitude of genetic risk factors involved, and the risk allele frequencies, large sample sets are often needed to obtain meaningful data. In the case of PD, assuming a recurrence risk for siblings (λsibs) of around 3, the number of affected sibling pairs (ASPs) needed to have enough power to detect linkage at 0.05 genome-wide significance level is predicted to be in the order of 200-300, assuming a single major genetic effect. The number of ASP needed is much higher if many loci are involved.37,38
Modeling the disease definition and phenotype boundaries is another critical issue in PD genetics. Models with narrower or broader disease definitions can be used.
The statistical corrections needed to declare significant linkage using multiple comparisons when several clinical models using the same dataset, or when subsets are analyzed after stratification of the whole dataset are still debated. Replication of positive findings between different datasets is therefore important in order to identify genuine linkages. Furthermore, regions found using these approaches are usually very large (because ASPs are closely related so the region shared IBD is large) and therefore, follow-up studies to pinpoint the location of the relevant gene are difficult.
The results of four genome-wide scans in families with affected relative pairs (ARP) with classical PD have been published so far (Table 2).7,36,39,40 Moreover, two of the same datasets have been reanalyzed in search of modifiers of PD onset age.8,41 Comparing the results of these studies is not straightforward, because studies differ in clinical and genetic designs, sample size, density of markers typed, and especially in statistical analyses. Significant linkage was detected to a new region on chromosome 1p32 (PARK10) in one scan,7 and to the parkin gene locus on 6q (PARK2) in another scan;40 moreover, the region on 1p32 was significantly linked to PD onset age in another study (fig. 4).8
A list of additional regions has been generated, with values of LODs ranging from 1 to around 2 (fig. 4). These findings represent nonsignificant, “interesting” or “suggestive” linkage signals that need to be further evaluated in future studies.
Several of these signals are nonconsistent across the studies, and they might be false positive results. However, the consistency of findings for a few regions suggests that these might represent additional genuine linkages, though the power of the separate studies was insufficient to achieve statistical significance.
A first study (the GenePD study) performed on 113 ASPs yielded no significant results, probably because of the limited sample size.39
A second study was performed on a slightly larger sample of 174 families with ≥1 ARPs, including 184 ASPs and 75 ARPs of other type (cousins, parent-child, avuncular).40 Again, in the overall dataset, no significant linkage was found. However, within a subset of 18 families containing at least 1 PD case with onset ≤40 years (early-onset families), significant evidence for linkage (LOD>5) was detected at the PARK2 locus (parkin). Notably, no signal to the PARK2 region was detected by the analysis on the overall dataset, highlighting the potential pitfalls in linkage analysis of multiple families with a genetically heterogeneous disease. A third study (Parkinson Study Group) analyzed 182 families with ASPs.36 In order to reduce genetic heterogeneity, 22 families with parkin gene mutations were excluded from the analysis. Moreover, affection status was modeled according to a narrower (model I) and a broader PD definition (modeI II). Analyses under model I yielded no significant LOD scores but reanalysis according to the model II (broader PD definition) increased the LODs at regions on 2q and Xq (LODs 2.5 and 2.7 respectively). The finding on chromosome 2q36-q37 was followed up in a larger dataset and now reached significance.42
The fourth study was performed in the genetically isolated population of Iceland.7
Genetically isolated populations are increasingly recognized as a powerful framework for research aimed at the identification of genetic factors underlying complex disorders.43 Since isolated populations originate from a limited number of founders, they are characterized by a simpler genetic make-up compared to the general population; this can greatly facilitate the identification of genes for complex diseases, such as PD. The caveat is that genetic determinants identified in genetic isolates might not be relevant in the general population.
A significant familial aggregation of PD was previously shown in Iceland.44 Using a population-based approach, fifty-one families were ascertained with ≥1 ARPs with PD, yielding a total of 117 patients. Nonparametric, model-free analysis generated significant evidence for linkage to a region on chromosome 1p32 (max LOD=4.9), and this locus was termed PARK10.7 In this dataset, maximizing the LODs under a variety of parametric models (dominant, additive, multiplicative) did not allow a particular model of inheritance to be delineated. However, calculations on the basis of the observed vs. expected alleles shared IBD in the dataset, the modeled disease allele frequencies, and the observed sibling relative risks in Iceland, it was estimated that this locus alone could account for a substantial fraction of the familial aggregation of PD in this population.7 While this is an important significant linkage finding, its relevance for the other populations remains unknown. None of the previous three genome-wide scans had detected linkage to 1p32.36,39,40
Among the “interesting” regions detected across the four genome-wide scans, the most consistent appear those on chromosomes 5q, 9q, 17q, and Xq (fig. 4).
These regions are very large and contain many genes, including obvious candidates: the gene encoding synphilin-1, an alpha-synuclein interactor (on 5q),45 the gene encoding dopamine β-hydroxylase, and DYT1, the gene causing generalized torsion dystonia (on 9q),46,47 and the gene for a monogenic, X-linked dystonia-parkinsonism form (on Xq).48 The peak detected on 17q is close to the gene for microtubule associated protein tau, an important locus for neurodegeneration and a known risk factor for progressive supranuclear palsy and PD.49,50
The NR4A2 (NURR1) gene, located at 2q22-q23, encodes a transcription factor important for the genesis and maintenance of dopaminergic neurons.51 Two mutations in the non_coding region of NR4A2 were recently reported in families with late-onset PD.52 NR4A2 maps more than 70 cM centromerically to the linkage peak detected in the study of Pankratz et al.36 Despite its intriguing functional links to PD, NR4A2 is therefore not a likely candidate for the reported 2q36-q37 region.
All previous studies were designed to identify genetic determinants of PD risk. However, age at disease onset (AAO) might also be genetically controlled, and finding such genetic determinants or modifiers of onset age might have important implications in order to delay the onset of disease. In this approach, the available information on AAO of patients and age at examination of unaffected family members can be used to model AAO as a quantitative trait locus (QTL), which is amenable for linkage analysis. Two groups have recently reanalyzed their datasets using this approach.8,41
The first study on PD onset age as a QTL8 reanalyzed the dataset published in Scott et al. 2001. According to these results, the estimated heritability of AAO for PD is high (62%), indicating that strong genetic determinants for PD onset age exist. Significant evidence for linkage of AAO was found for markers in the region of chromosome1p32 (LOD=3.41). This region overlaps with the PARK10 region found in the Icelandic study.7 Additional regions yielding LODs between 1.4 and 1.9 were found on chromosomes 5q, 6p, 10q, 13q, 17p, 20p and 22q (fig.4). Moreover, when the PD dataset was analyzed together with a dataset of AD families, evidence was found for the existence of a genetic determinant of onset age for both diseases on chromosome 10q.8 Although most of the evidence came from the group of AD patients, which was significantly larger then the group of PD patients, this finding is still intriguing since clinical, biochemical and pathological overlaps between PD and AD have been documented, and it is therefore conceivable that common genetic factors control the pathogenesis and/or the progression in both the diseases. Moreover, together with the results of the Icelandic study, the detection of linkage on 1p32 confirms that an important genetic determinant of PD risk and/or onset age resides in this region.
The other study41 reanalyzed the dataset of the GenePD study and found suggestive evidence (LODs between 1.8 to 2.2) for linkage of AAO to chromosome 2p, 9q, 20q, and 21 (fig. 4). The region on 2p coincides with PARK3, a locus for autosomal dominant, late-onset PD,53 whereas the region on 9q has also been detected in linkage to affection status in the GenePD and in another screen (fig. 4).
In addition to linkage to PARK3, evidence for association was also observed between a specific allele of D2S1394 (one of the markers within PARK3) and AAO.41
In addition to 2p (PARK3) and 1p32 (PARK10), other regions like the one on 5q and 9q have been detected by different studies searching for linkage to disease status (genetic risk) and for linkage to AAO (fig. 4). It is well possible that current modeling and statistical methods lead to detection of genetic determinants of AAO when studying genetic risk, and vice versa.
With the exception of PARK3, the current results of the genome scans suggest that the genes implicated in the known Mendelian forms are not major loci for common forms of PD. However, the failure to detect significant linkage, and the inconsistency of most peaks across different scans, suggest that most of these studies were underpowered. A still unknown major locus for susceptibility to common forms of PD could have escaped detection just due to power limitations. The results are also compatible with the existence of many weaker genetic risk factors, which might also differ in the different studies, particularly if genetically isolated populations are targeted. In the light of the results of screens published so far for late-onset PD, targeting a genetically isolated population has been more rewarding than scanning series of affected-pairs from the general populations.
Conclusions
Classical linkage approaches to monogenic forms of PD have been most fruitful in the last few years, but it is anticipated that more and more genome-wide studies will be performed in the coming years in the common form of the disease, using families with affected-pairs, and cohorts of isolated cases from genetically isolated populations. The technology of DNA microarrays (DNA chips) is also anticipated to contribute soon to the identification of PD-related genes.
A high level of etiological complexity likely exists for PD; in this scenario, genetic approaches to isolated populations or otherwise-defined more homogeneous groups is convenient in order to try and dissect the complexity of the problem.
During the last year the finished sequence of the human genome has been released. At the beginning of the post-genomic era, there are motifs to believe that the genome-wide approach will also succeed in deciphering the code of PD.
Acknowledgements
We thank the National Parkinson Foundation (USA), the Parkinson's Disease Foundation (USA), and the Prinses Beatrix Fonds (Netherlands) for their support.
References
- 1.
- Lang AE, Lozano AM. Parkinson's disease. First of two parts. N Engl J Med. 1998;339:1044–1053. [PubMed: 9761807]
- 2.
- Dawson TM, Dawson VL. Rare genetic mutations shed light on the pathogenesis of Parkinson disease. J Clin Invest. 2003;111:145–151. [PMC free article: PMC151882] [PubMed: 12531866]
- 3.
- Cookson MR. Pathways to parkinsonism. Neuron. 2003;37:7–10. [PubMed: 12526767]
- 4.
- Valente EM, Bentivoglio AR, Dixon PH. et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001;68:895–900. [PMC free article: PMC1275643] [PubMed: 11254447]
- 5.
- van DuijnCM, Dekker MC, Bonifati V. et al. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet. 2001;69:629–634. [PMC free article: PMC1235491] [PubMed: 11462174]
- 6.
- Hampshire DJ, Roberts E, Crow Y. et al. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet. 2001;38:680–682. [PMC free article: PMC1734748] [PubMed: 11584046]
- 7.
- Hicks AA, Petursson H, Jonsson T. et al. A susceptibility gene for late-onset idiopathic Parkinson's disease. Ann Neurol. 2002;52:549–555. [PubMed: 12402251]
- 8.
- Li YJ, Scott WK, Hedges DJ. et al. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet. 2002;70:985–993. [PMC free article: PMC379130] [PubMed: 11875758]
- 9.
- Bonifati V, Rizzu P, van Baren MJ. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. [PubMed: 12446870]
- 10.
- Bonifati V, Breedveld GJ, Squitieri F. et al. Localization of autosomal recessive early-onset parkinsonism to chromosome 1p36 (PARK7) in an independent dataset. Ann Neurol. 2002;51:253–256. [PubMed: 11835383]
- 11.
- Taira T, Takahashi K, Kitagawa R. et al. Molecular cloning of human and mouse DJ-1 genes and identification of Sp1-dependent activation of the human DJ-1 promoter. Gene. 2001;263:285–292. [PubMed: 11223268]
- 12.
- Nagakubo D, Taira T, Kitaura H. et al. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun. 1997;231:509–513. [PubMed: 9070310]
- 13.
- Hod Y, Pentyala SN, Whyard TC. et al. Identification and characterization of a novel protein that regulates RNA-protein interaction. J Cell Biochem. 1999;72:435–444. [PubMed: 10022524]
- 14.
- Welch JE, Barbee RR, Roberts NL. et al. SP22: a novel fertility protein from a highly conserved gene family. J Androl. 1998;19:385–393. [PubMed: 9733139]
- 15.
- Wagenfeld A, Gromoll J, Cooper TG. Molecular cloning and expression of rat contraception associated protein 1 (CAP1), a protein putatively involved in fertilization. Biochem Biophys Res Commun. 1998;251:545–549. [PubMed: 9792810]
- 16.
- Takahashi K, Taira T, Niki T. et al. DJ-1 positively regulates the androgen receptor by impairing the binding of PIASx alpha to the receptor. J Biol Chem. 2001;276:37556–37563. [PubMed: 11477070]
- 17.
- Kotaja N, Karvonen U, Janne OA. et al. PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol Cell Biol. 2002;22:5222–5234. [PMC free article: PMC139781] [PubMed: 12077349]
- 18.
- Mitsumoto A, Nakagawa Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic Res. 2001;35:885–893. [PubMed: 11811539]
- 19.
- Mitsumoto A, Nakagawa Y, Takeuchi A. et al. Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radic Res. 2001;35:301–310. [PubMed: 11697128]
- 20.
- de NobelH, Lawrie L, Brul S. et al. Parallel and comparative analysis of the proteome and transcriptome of sorbic acid-stressed Saccharomyces cerevisiae. Yeast. 2001;18:1413–1428. [PubMed: 11746603]
- 21.
- Trotter EW, Kao CM, Berenfeld L. et al. Misfolded proteins are competent to mediate a subset of the responses to heat shock in Saccharomyces cerevisiae. J Biol Chem. 2002;277:44817–44825. [PubMed: 12239211]
- 22.
- Giasson BI, Ischiropoulos H, Lee VM. et al. The relationship between oxidative/nitrative stress and pathological inclusions in Alzheimer's and Parkinson's diseases. Free Radic Biol Med. 2002;32:1264–1275. [PubMed: 12057764]
- 23.
- Zoghbi HY, Botas J. Mouse and fly models of neurodegeneration. Trends Genet. 2002;18:463–471. [PubMed: 12175807]
- 24.
- Hyun DH, Lee M, Hattori N. et al. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J Biol Chem. 2002;277:28572–28577. [PubMed: 12034719]
- 25.
- Hashimoto M, Hsu LJ, Rockenstein E. et al. alpha-Synuclein protects against oxidative stress via inactivation of the c-Jun N-terminal kinase stress-signaling pathway in neuronal cells. J Biol Chem. 2002;277:11465–11472. [PubMed: 11790792]
- 26.
- Valente EM, Bentivoglio AR, Cassetta E. et al. DYT13, a novel primary torsion dystonia locus, maps to chromosome 1p36.13-36.32 in an Italian family with cranial-cervical or upper limb onset. Ann Neurol. 2001;49:362–366. [PubMed: 11261511]
- 27.
- Valente EM, Brancati F, Ferraris A. et al. PARK6-linked parkinsonism occurs in several European families. Ann Neurol. 2002;51:14–18. [PubMed: 11782979]
- 28.
- Khan NL, Valente EM, Bentivoglio AR. et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: An 18F-dopa PET study. Ann Neurol. 2002;52:849–853. [PubMed: 12447943]
- 29.
- Bentivoglio AR, Cortelli P, Valente EM. et al. Phenotypic characterisation of autosomal recessive PARK6-linked parkinsonism in three unrelated Italian families. Mov Disord. 2001;16:999–1006. [PubMed: 11748730]
- 30.
- Lucking CB, Durr A, Bonifati V. et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. French Parkinson's Disease Genetics Study Group. N Engl J Med. 2000;342:1560–1567. [PubMed: 10824074]
- 31.
- Dekker M, Bonifati V, van Swieten J. et al. Clinical features and neuroimaging of PARK7-linked parkinsonism. Mov Disord. 2003;18:751–757. [PubMed: 12815653]
- 32.
- Najim al-DinAS, Wriekat A, Mubaidin A. et al. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand. 1994;89:347–352. [PubMed: 8085432]
- 33.
- Funayama M, Hasegawa K, Kowa H. et al. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. 2002;51:296–301. [PubMed: 11891824]
- 34.
- Hasegawa K, Kowa H. Autosomal dominant familial Parkinson disease: older onset of age, and good response to levodopa therapy. Eur Neurol. 1997;38:39–43. [PubMed: 9276200]
- 35.
- Scott WK, Grubber JM, Conneally PM. et al. Fine mapping of the chromosome 12 late-onset Alzheimer disease locus: Potential genetic and phenotypic heterogeneity. Am J Hum Genet. 2000;66:922–932. [PMC free article: PMC1288173] [PubMed: 10712207]
- 36.
- Pankratz N, Nichols WC, Uniacke SK. et al. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet. 2002;71:124–135. [PMC free article: PMC384969] [PubMed: 12058349]
- 37.
- Risch N. Evolving methods in genetic epidemiology. II. Genetic linkage from an epidemiologic perspective. Epidemiol Rev. 1997;19:24–32. [PubMed: 9360899]
- 38.
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. [PubMed: 8801636]
- 39.
- DeStefano AL, Golbe LI, Mark MH. et al. Genome-wide scan for Parkinson's disease: The GenePD Study. Neurology. 2001;57:1124–1126. [PubMed: 11571351]
- 40.
- Scott WK, Nance MA, Watts RL. et al. Complete genomic screen in Parkinson disease: Evidence for multiple genes. JAMA. 2001;286:2239–2244. [PubMed: 11710888]
- 41.
- DeStefano AL, Lew MF, Golbe LI. et al. PARK3 influences age at onset in Parkinson disease: A genome scan in the GenePD study. Am J Hum Genet. 2002;70:1089–1095. [PMC free article: PMC447587] [PubMed: 11920285]
- 42.
- Pankratz N, Nichols WC, Uniacke SK. et al. Significant Linkage of Parkinson Disease to Chromosome 2q36-37. Am J Hum Genet. 2003;72:1053–1057. [PMC free article: PMC1180337] [PubMed: 12638082]
- 43.
- Heutink P, Oostra BA. Gene finding in genetically isolated populations. Hum Mol Genet. 2002;11:2507–2515. [PubMed: 12351587]
- 44.
- Sveinbjornsdottir S, Hicks AA, Jonsson T. et al. Familial aggregation of Parkinson's disease in Iceland. N Engl J Med. 2000;343:1765–1770. [PubMed: 11114315]
- 45.
- Engelender S, Kaminsky Z, Guo X. et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110–114. [PubMed: 10319874]
- 46.
- Craig SP, Buckle VJ, Lamouroux A. et al. Localization of the human dopamine beta hydroxylase (DBH) gene to chromosome 9q34. Cytogenet Cell Genet. 1988;48:48–50. [PubMed: 3180847]
- 47.
- Ozelius LJ, Hewett JW, Page CE. et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. [PubMed: 9288096]
- 48.
- Wilhelmsen KC, Weeks DE, Nygaard TG. et al. Genetic mapping of Lubag (X-linked dystonia-parkinsonism) in a Filipino kindred to the pericentromeric region of the X chromosome. Ann Neurol. 1991;29:124–131. [PubMed: 1672807]
- 49.
- Heutink P. Untangling tau-related dementia. Hum Mol Genet. 2000;9:979–986. [PubMed: 10767321]
- 50.
- Martin ER, Scott WK, Nance MA. et al. Association of single-nucleotide polymorphisms of the tau gene with late-onset Parkinson disease. JAMA. 2001;286:2245–2250. [PMC free article: PMC3973175] [PubMed: 11710889]
- 51.
- Le W, Conneely OM, Zou L. et al. Selective agenesis of mesencephalic dopaminergic neurons in Nurr1-deficient mice. Exp Neurol. 1999;159:451–458. [PubMed: 10506516]
- 52.
- Le WD, Xu P, Jankovic J. et al. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2003;33:85–89. [PubMed: 12496759]
- 53.
- Gasser T, Muller-Myhsok B, Wszolek ZK. et al. A susceptibility locus for Parkinson's disease maps to chromosome 2p13. Nat Genet. 1998;18:262–265. [PubMed: 9500549]
- 54.
- Polymeropoulos MH, Lavedan C, Leroy E. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. [PubMed: 9197268]
- 55.
- Kitada T, Asakawa S, Hattori N. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. [PubMed: 9560156]
- 56.
- Farrer M, Gwinn-Hardy K, Muenter M. et al. A chromosome 4p haplotype segregating with Parkinson's disease and postural tremor. Hum Mol Genet. 1999;8:81–85. [PubMed: 9887334]
- 57.
- Leroy E, Boyer R, Auburger G. et al. The ubiquitin pathway in Parkinson's disease. Nature. 1998;395:451–452. [PubMed: 9774100]
Figures
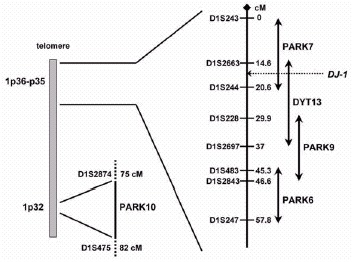
Figure 1
Chromosome 1p map illustrating the position of the novel PD-related loci.
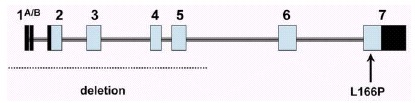
Figure 2
Genomic structure of the DJ-1 gene. The dark and light boxes indicate noncoding and coding exonic sequence, respectively. Indicated are the two mutations identified so far.
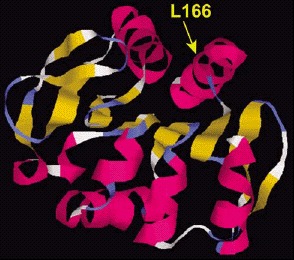
Figure 3
Structural model of the DJ-1 protein. The dark and light ribbons indicate regions with α-helix and β-sheet structure, respectively. The position of the amino acid mutated in one PD family (L166) is shown.

Figure 4
Overview of results from the genome-wide screens in common forms of PD. The black, shaded, and grey boxes indicate regions with LOD scores values >3, >2, and >1, respectively.
Tables
Table 1Current catalogue of genes and loci for PD
| Locus* | OMIM | Map Position | Gene | Inheritance | Pathology | Ref. |
|---|---|---|---|---|---|---|
| PARK1 | #168601 | 4q21-q23 | α-syncluein | dominant, high penetrance | LB+ | 54 |
| PARK2 | #600116 | 6q25-q27 | parkin | recessive | mostly LB negative | 55 |
| PARK3 | 602604 | 2p13 | unknown | dominant-incomplete penetrance | LB+ | 53 |
| PARK4 | 605543 | 4p15 | unknown | dominant, high penetrance | LB+ | 56 |
| PARK5 | 191342 | 4p14 | UCH-L1 | dominant | unknown | 57 |
| PARK6 | 605909 | 1p35-p36 | unknown | recessive | unknown | 4 |
| PARK7 | #606324 | 1p36 | DJ-1 | recessive | unknown | 9 |
| PARK8 | 607060 | 12p11-q13 | unknown | dominant, incomplete penetrance | LB negative | 33 |
| PARK9 | pending | 1p36 | unknown | recessive | unknown | 6 |
| PARK10 | 606852 | 1p32 | unknown | non-Mendelian | unknown | 7 |
| pending | 601828 | 2q22-q23 | NR4A2(NURR1) | dominant(?) | unknown | 52 |
| pending | pending | 2q36-q37 | unknown | non-mendelian | unknown | 42 |
* Named according to HUGO Gene Nomenclature Committee (http://www.gene.ucl.ac.uk/nomenclature).
Table 2The genome-wide screens performed in the common forms of PD
| Study | Dataset | Sample Size | Marker Density | Software Packages |
|---|---|---|---|---|
| OBJECT: Determinants of PD risk | ||||
| De Stefano et al (Neurology 2001) | sib-pairs (GenePD study) | 113 ASP (102 families) | 11 cM (339 markers) | GENEHUNTER |
| Scott et al (JAMA 2001) | families with ≥2 aff. relatives | 260 ARP (174 families) | 10 cM (344 markers) | VITESSE/HOMOG (par.) GENEHUNTER-PLUS |
| Pankratz et al (Am J Hum Genet 2002) | sib-pairs (Parkinson Study Group) | 170 ASP (160 families) | 8.6 cM (400 markers) | MAPMAKER-SIBS |
| Hicks et al (Ann Neurology 2002) | isolated population (Iceland) | 117 patients (51 families) | <5 cM (781 markers) | ALLEGRO |
| OBJECT: Determinants of PD onset age (AAO) | ||||
| Li et al (Am J Hum Genet 2002) | families with ≥2 aff. relatives | see Scott et al 2001 | see Scott et al 2001 | SOLAR |
| De Stefano et al (Am J Hum Genet 2002) | sib-pairs (Gene PD study) | see De Stefano et al, 2001 | see De Stefano et al 2001 | GENEHUNTER |