

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Collaborating Centre for Nursing and Supportive Care (UK). Irritable Bowel Syndrome in Adults: Diagnosis and Management of Irritable Bowel Syndrome in Primary Care [Internet]. London: Royal College of Nursing (UK); 2008 Feb. (NICE Clinical Guidelines, No. 61.)

Update information March 2017 Recommendation 1.1.1.2 in the short version was updated by NICE with more recent guidance on recognition and referral for suspected cancer. Recommendation 1.1.1.3 in the short version was removed as it was no longer needed after the changes to recommendation 1.1.1.2. February 2015 NICE has made new recommendations relating to the clinical management (dietary and lifestyle advice, and pharmacological therapy) of people with IBS. The recommendations and evidence in sections 3, 7.6, 8.4, 8.5.2 and 9.1 of this guideline that have been highlighted in grey have been stood down and replaced. New recommendations on dietary and lifestyle advice, and pharmacological therapy, can be found in the irritable bowel syndrome in adults update CG61.1. September 2012 A recommendation in this guideline (see pages 28 and 37) has been partially updated by recommendation 1.1.2.1 in 'Ovarian cancer' (NICE clinical guideline 122, 2011).

Irritable Bowel Syndrome in Adults: Diagnosis and Management of Irritable Bowel Syndrome in Primary Care [Internet].
Show detailsClinical Questions
- Are antispasmodics effective in managing IBS symptoms?
- Are laxatives effective in the management of IBS?
- Are anti-motility agents effective in symptom control in IBS?
- Do tricyclics and SSRI’s have a role in the management of IBS symptoms?
BACKGROUND
The pharmacological management of IBS can provide clinicians with a major therapeutic challenge. People with IBS may present with a multi-symptom profile and it is unlikely that all patients will respond in the same way to the same single agent. There have been no new drugs specifically developed for the treatment of IBS in the last twenty years and the quality of the trials in the majority of pharmacological agents currently available is variable and often conducted on secondary populations. The drug management strategy should be based on the nature and severity of the symptoms and individual or combinations of medication directed at the predominant symptom/s. Irritable bowel syndrome can present with pain, constipation, or diarrhoea. Antispasmodic, TCA and SSRI drugs may relieve IBS pain. Antimotility drugs may relieve diarrhoea. Opioids with a central action such as codeine are better avoided because of the risk of dependence. Laxatives may be needed to relieve constipation. It is important to be sure that the patient is constipated. People who complain of constipation need to understand that bowel habit can vary considerably in frequency without doing harm. Some people tend to consider themselves constipated if they do not have a bowel movement each day. Misconceptions about ‘normal’ bowel habits have led to excessive or inappropriate laxative use. Laxative abuse may lead to hypokalaemia.
In some people with IBS there may be important psychological aggravating factors which respond to reassurance and possibly specific treatment e.g. with an antidepressant.
Antispasmodics
The abdominal pain experienced by people with IBS may be a result of irregular and intermittent intestinal contractions along the length of the colon. This may lead to symptoms of abdominal pain, bloating and gas. Pain is most common after a meal and may last for several hours.
Antispasmodics can be separated into two main categories: antimuscarinics, and smooth muscle relaxants. Antimuscarinics reduce intestinal motility; smooth muscle relaxants directly relax intestinal smooth muscle. The use of antispasmodics is primarily to relax the smooth muscles of the gut, helping to prevent or relieve the painful cramping spasms in the intestines. They are typically taken 30 to 45 minutes before meals.
Antimotility agents
Diarrhoea is associated with alterations of fluid and electrolyte movement in either the small intestine or the colon. This can be due to decreased intestinal absorption, altered intestinal motility, or increased intestinal secretions (e.g. due to bacterial enterotoxins or laxatives). Antimotility agents are used to manage acute or chronic diarrhoea or exacerbations of chronic diarrhoea and work by altering one or more of these mechanisms.
Antimotility agents for IBS can be separated into four main categories: codeine phosphate; co-phenotrope (mixture of diphenoxylate hydrochloride and atropine sulphate in the mass proportions 100 parts to 1 part); loperamide and morphine-containing preparations. Prolonged codeine use can lead to dependency. Loperamide is considered especially useful as it tends to increase anal sphincter tone.
Laxatives
Laxatives can be separated into four main categories: bulk forming laxatives; stimulant laxatives; faecal softeners and osmotic laxatives. Bulk-forming laxatives relieve constipation by increasing faecal mass, which stimulates peristalsis; adequate fluid intake should be maintained to avoid intestinal obstruction. Stimulant laxatives work by increasing intestinal motility, but they often cause abdominal cramps. Faecal softeners may lubricate the passage of stools and/or soften them. Osmotic laxatives increase the amount of water in the large bowel, either by drawing fluid from the body into the bowel or by retaining the fluid with which they were administered. The route of adminstration for laxatives may be oral or rectal. Laxatives can be used in two ways: as short-term rescue medication or as longer-term maintenance treatment. There is no evidence that long term laxative use damages the bowel.
Tricyclics and Antidepressants
Since their introduction approximately fifty years ago, antidepressants have been used in a variety of gastrointestinal (GI) conditions. In the last twenty years antidepressants have been increasingly used in the treatment of functional GI disorders such as IBS. The prevalence of anxiety and depressive disorders is high in patients with severe and/or intractable IBS and may be present to some degree in all IBS patients. Antidepressants appear have an analgesic effect separate to their antidepressant effect. Visceral pain syndromes including IBS may be effectively treated by a range of therapies, including antidepressants that modulate the interactions between the central and enteric nervous systems. Tricyclics also have a peripheral anticholinergic action in addition to their central analgesic and antidepressant actions.
Antidepressants can be divided into three major classes: tricyclics and related antidepressants; selective serotonin re-uptake inhibitors (SSRIs), and monoamine oxidase inhibitors (MAOIs). There are other antidepressants that do not fit easily into these categories: Duloxetine (Cymbalta); Flupentixol (Fluanxol); Mirtazapine (Zispin Soltab); Reboxetine (Edronax); Tryptophan (Optimax), and; Venlafaxine (Efexor).
8.1. Laxatives
SELECTION CRITERIA
The selection criteria described in the general methodology section were used, but some were specific to the laxatives review and are reported below.
Types of studies
For longer-term studies, the GDG decided that the washout period for this review should be at least two weeks. Trials with no washout were not included in the analysis and trials with one week washout were considered if there was no other information. Crossover studies are not appropriate for short-term (rescue) medication.
Types of participants
For this review, participants with IBS were included, but the review was extended to include people with simple constipation as well because the same drugs are used. Studies in these participants were regarded as indirect as far as population was concerned.
Types of intervention
Studies were to include the following interventions:
- Bulk-forming laxatives, which, as a class, include some dietary fibres (see fibres review), but here we consider only non-dietary bulk forming agents:
- Ispaghula husk (trade names: Fibrelief®; Fybogel®; Isogel®; Ispagel Orange®; Regulan®)
- Methylcellulose (trade name: Celevac®)
- Sterculia (trade names: Normacol®; Normacol Plus®).
- Stimulant laxatives:
- Biascodyl (trade name: *Dulco-lax®), given as either oral tablets or rectal suppositories
- Docusate sodium (synonym: dioctyl sodium sulphosuccinate; trade names: Dioctyl® (oral); Docusol® (oral); Norgalax Micro-enema® (rectal))
- Glycerol (synonym: glycerine), given as rectal suppositories
- Senna (non proprietary tablets; trade names: Senokot® granules; Manevac® granules (senna fruit 12.4%, ispaghula 54.2%)), given as an oral preparation
- Faecal softeners:
- Arachis oil (Trade name: Fletchers’Arachis Oil Retention Enema®), given as a rectal preparation
- Liquid paraffin (Liquid Paraffin Oral Emulsion, BP), given as oral preparation.
- Osmotic laxatives:
- Lactulose (trade names include: Duphalac®; Lactugal®; Regulose®), given as oral solution
- Macrogols (synonyms: polyethylene glycols, PEG; trade names: Idrolax® (oral powder, PEG 4000); Movicol® (oral powder, PEG 3350); Movicol®-Half (oral powder, PEG 3350))
- Magnesium salts (Magnesium Hydroxide Mixture, BP, oral aqueous suspension; Liquid Paraffin and Magnesium Hydroxide Oral Emulsion, BP (oral aqueous suspension), Magnesium Sulphate (Epsom Salts); trade name: Milpar®)
- Sodium phosphate – this is not in the BNF but is in routine use and so was included by the GDG.
The following comparisons were included:
- Laxative versus placebo (or nothing)
- Laxative type 1 versus type 2
- Laxative dose 1 versus dose 2
- Laxative + another intervention versus the other intervention alone
- Laxative route of delivery 1 versus route 2
- Duration of treatment 1 versus duration 2.
NB: In spite of the large placebo effect associated with IBS, comparisons with no treatment are included.
The laxatives review was concerned with both longer-term maintenance treatment and short-term symptom relief.
The GDG decided that there should be a minimum duration of treatment of four weeks for maintenance in this review. Maintenance studies of shorter durations were not included in the analysis.
Subgroup analyses
We carried out subgroup analyses by type of laxative (bulk forming laxatives; stimulant laxatives; faecal softeners, and; osmotic laxatives); dose; route of delivery (oral, rectal), and; duration of intervention.
SEARCH STRATEGY FOR IDENTIFICATION OF STUDIES
The initial search identified a Cochrane Review (Quartero 2005, Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome). Searches were partly based on the terms in this review. Searches were performed on the following core databases: MEDLINE, EMBASE, CINAHL and The Cochrane Library (1966 to current day with guidance from the GDG).
Additional databases were not searched for this review. For this review, the search was extended to cover the population with simple constipation as well as IBS. The search strategies are listed in Appendix B.
The Cochrane review identified 11 studies, ten of which are included in the fibres review. The remaining study (Piai 1987) used a cellulose material, glucomannan, which is not used in the UK. The titles and abstracts identified by the NCC search strategy were assessed and fifty studies were retrieved in full. The reference lists for each of the retrieved studies were inspected for further potential papers, but none were identified. The 38 excluded studies are listed in Appendix E, along with reasons for exclusion. Searches were updated to June 2007 and a further two papers were identified and obtained from the authors with some further information (Wulkow 2007; Kienzle-Horn 2007).
DESCRIPTION OF STUDIES INCLUDED IN THE REVIEW
Study Design
There were four crossover studies (Cleveland 2001; Connolly 1975; Quah 2006; Sobhani 1996) in which participants were allocated to receive both the intervention and control treatments during the course of the study, in a random order. Two of these studies (Cleveland 2001; Sobhani 1996) had either no washout period or it was not reported, in which case this was assumed to be none. The other two studies had a washout period of one week. The GDG had specified a washout period of two weeks minimum, but would consider the two one-week studies if there was no other data. No crossover studies reported first-period results only or individual patient data.
The remaining 15 studies had a parallel design (Attar 1999; Bouhnik 2004; Chaussade 2003; Corazziari 1996; Corazziari 2000; Dettmar 1998; DiPalma 2000; Hamilton 1988; Kienzle-Horn 2006; Kienzle-Horn 2007; Marlett 1987; Medoff 2004; Rouse 1991; Wang 2004; Wulkow 2007).
The GDG had specified a minimum treatment period of four weeks for each intervention in the maintenance studies. Two studies had a treatment duration of one week (Connolly 1975; Marlett 1987); one had ten days (Hamilton 1988); three had two weeks (Cleveland 2001; DiPalma 2000; Wang 2004); one had three weeks (Sobhani 1996). All these studies were transferred to the excluded studies table.
The remaining studies had durations of four weeks (Attar 1999; Bouhnik 2004; Chaussade 2003; Dettmar 1998; Kienzle-Horn 2007; Medoff 2004; Quah 2006 (1-week washout crossover); Rouse 1991); eight weeks (Corazziari 1996), and; 20 weeks (Corazziari 2000).
Two studies investigated the use of laxatives for acute constipation (Kienzle-Horn 2006; Wulkow 2007).
Twelve studies were therefore included in the analysis (11 parallel and one 1-week washout crossover trial: Attar 1999; Bouhnik 2004; Chaussade 2003; Corazziari 1996; Corazziari 2000; Dettmar 1998; Kienzle-Horn 2006; Kienzle-Horn 2007; Medoff 2004; Quah 2006; Rouse 1991; Wulkow 2007).
One of the remaining studies had more than two arms: Chaussade (2003) compared four PEG interventions; there were thus 14 comparisons in the laxatives review. The rest of the description of studies will focus on these studies/comparisons.
Two were conducted in the UK (Dettmar 1998 and Rouse 1991); one in sites in the UK and France (Attar 1999); seven in the rest of Europe, one in each of the USA and China.
Setting: Seven studies took place in primary care (Bouhnik 2004; Chaussade 2003; Dettmar 1998; Kienzle-Horn 2006; Medoff 2004; Rouse 1991; Wulkow 2007); and five were in secondary care (Attar 1999 (with 31% from geriatric institutions); Corazziari 1996 Corazziari 2000; Kienzle-Horn 2007; Quah 2006).
The majority of studies (7/12) had fewer than 100 patients, with two having 25 or fewer in the intervention arm (Corazziari 1996; Medoff 2004). Two studies had more than 200 patients in total (Chaussade 2003; Dettmar 1998).
Funding: Six studies had some industry sponsorship: Bouhnik (2004) was sponsored by Solvay Pharmaceuticals, manufacturers of lactulose; Chaussade (2003) was supported by a grant from by Hoffmann La Roche, the manufacturers of PEG 3350, and; the Dettmar (1998) authors were from Reckitt & Colman, manufacturers of Fybogel. Kienzle-Horn (2006), Kienzle-Horn (2007) and; Wulkow (2007) were all funded by Boehringer-Ingelheim GmbH, manufacturers of Bisacodyl, and Sodium Picosulphate.
Population
- Only one study (Medoff 2004) definitely included patients with IBS: 7/43 patients had a diagnosis of constipation predominant IBS, but separate results were not reported. All the other studies stated they had patients with simple constipation, usually defined as 3 or less, or 2 or less bowel movements per week. Most studies defined a minimum period of constipation symptoms, ranging from 3 weeks (Rouse 1991) to 12 months (Corazziari 1996; Corazziari 2000). The GDG suspected, however, that many of these studies may have had the classification of ‘simple constipation’ because this was the primary symptom being treated by laxatives, rather than the only symptom. Indeed, they thought it was quite likely that the patients had IBS. This was further investigated. Communication with the authors of one study (Kienzle-Horn 2007) revealed that abdominal pain was not excluded and the author believed that some patients entering the studies would have had IBS if they had been checked for it by their physician. Seven of these studies (Attar 1999; Bouhnik 2004; Corazziari 1996; Corazziari 2000; Chaussade 2003; Dettmar 1998; Rouse 1991) reported that some patients had pain and/or bloating. However, Dettmar (1998) and Rouse (1991) did not report sufficient duration to be defined as IBS. Two studies did not mention the incidence of pain or bloating before treatment (Kienzle-Horn 2006; Quah 2006) but the patients had had constipation for at least 3 months.
- In Attar (1999), 20% and 35% of the control group had pain and bloating respectively during the trial. The patients were stated to have had chronic idiopathic constipation for at least 3 months.
- In Bouhnik (2004), in the washout period, 45 and 53% had bloating at washout and 30 and 45% had pain for lactulose and PEG respectively. The patients were stated to have had chronic idiopathic constipation for at least 6 months.
- In Chaussade (2003), at baseline, the bloating score was ~ 3 points on a scale of 1 to 4 (considerable) and pain was 2.6. There was an implied use of the Rome II criteria for chronic idiopathic constipation, which the patients were to have had for at least 3 months
- In Corazziari (1996), in the run-in period 52–60% pts had pain and 84–91% had bloating. The patients were stated to have chronic non-organic constipation and had had this for at least 12 months.
- In Corazziari (2000), the pain and bloating scores were non zero, even after the patients had received PEG for 4 weeks. Chronic constipation was defined using Rome criteria and the patients had had chronic constipation for at least 12 months.
- Dettmar (1998) reported that the majority of the patients experienced abdominal symptoms, including pain, distension or flatulence. The patients were said to have ‘simple constipation’ and there were no details about the duration of constipation.
- In Rouse (1991) 53–54% patients in both groups had abdominal pain after seven days. Bloating was not mentioned. The patients were treated for chronic constipation, which they had had for at least 3 weeks.
The GDG concluded that all of these studies, with the exception of Dettmar (1998) and Rouse (1991) were likely to have some, if not all, patients with IBS. It was unclear if Kienzle-Horn (2006) and Quah (2006) included patients with IBS. Further details are given in the included studies table (Appendix C).
One study (Corazziari 2000) gave PEG electrolyte to patients for 4 weeks and then randomised only the responders (at least two bowel movements per week with no other defaecatory disturbances or more than three bowel movements per week) to PEG electrolyte or placebo.
The age range of participants across the studies was 18 to 89 years, with the mean age (where given) ranging from 42 to 58 years. All the studies had more women than men.
Interventions
The studies varied in the type of laxatives used:
- Two used stimulant laxatives:
- One bisacodyl (Kienzle-Horn 2006)
- One sodium picosulphate (Wulkow 2007).
- Eight had osmotic laxatives:
- Five polyethylene glycol (Attar 1999; Chaussade 2003; Corazziari 1996; Corazziari 2000; Bouhnik 2004)
- One sodium phosphate (Medoff 2004).
- One study (Dettmar 1998) allowed the patient any laxative (which was mainly lactulose) and also reported the lactulose patients as a subgroup.
Comparisons
The included studies covered the following comparisons:
- Four comparisons of laxatives versus placebo:
- Two gave stimulant laxatives (Kienzle-Horn 2006; Wulkow 2007 treatment for acute episodes)
- Two gave osmotic laxatives (Corazziari 1996; Corazziari 2000).
- Three studies compared a laxative with fibre:
- Two compared an osmotic laxative (lactulose) with fibre (ispaghula), (Quah 2006; Rouse 1991)
- One compared usual laxatives (mainly lactulose) with fibre (ispaghula) (Dettmar 1998).
- Seven comparisons of different types of laxative in the same class:
- Two studies (Attar 1999; Bouhnik 2004) compared lactulose with PEG electrolyte (Osmotic Laxatives)
- Four comparisons of PEG 3350 (Transipeg) plus electrolytes versus PEG 4000 without electrolytes (Forlax) (Chaussade 2003 x4) (Osmotic Laxatives)
- One comparison of Bisacodyl versus Sodium Picosulphate (Kienzle–Horn 2007).
- Three comparisons of different doses of an osmotic laxative:
- One comparison of 11.8g versus 5.9g PEG 3350 (Transipeg) plus electrolytes (Chaussade 2003)
- One comparison of 20g versus 10g PEG 4000 (Forlax) (Chaussade 2003)
- One comparison of 10.56g (mean) versus 6.84g (mean) sodium phosphate.
METHODOLOGICAL QUALITY
The results of the quality assessment for included trials are shown in Appendix D.
An adequate method of randomisation was reported in four studies (Attar 1999; Medoff 2004; Quah 2006; Wulrow 2007), all of which used a computer generated method. The other studies did not state the method.
Allocation concealment was reported in three studies (Attar 1999; Bouhnik 2004; Quah 2006), both of which reported an adequate method, in which the statistician prepared the list and the investigators were unaware of the allocation (Attar 1999) or by telephoning a central office (Bouhnik 2004; Quah 2006).
Six studies reported that the patients were blinded to the interventions (Chaussade 2003; Corazziari 1996; Corazziari 2000; Kienzle-Horn 2006; Kienzle-Horn 2007; Wulrow 2007); these included all the placebo controlled studies. The remaining studies stated that the patients were not blinded, or could not have been because of differences between drugs in appearance and taste.
Five studies (Attar 1999; Bouhnik 2004; Kienzle-Horn 2006; Kienzle-Horn 2007; Wulrow 2007) described an a-priori power calculation. All studies included in the review demonstrated baseline comparability of the groups, apart from one study which was not comparable at baseline (Medoff 2004) for rectal irritation, which was greater in the group receiving four tablets.
There was loss to follow-up in the majority of studies, and all but one had less than 20% dropouts. One study (Corazziari 2000) reported that more than 20% of patients in at least one arm (or overall) were not analysed or were lost to follow-up (attrition bias). In Corazziari (2000), for the first eight weeks 1/33 (3%) PMF and 4/37 (11%) placebo did not complete the period, but 10/33 (30%) PMF and 22/37 (59%) placebo did not complete the 20 weeks. Consequently, results at eight weeks only were taken for this study. In Quah (2006), 8/50 (22%) withdrew before receiving the interventions, then 3/21 (14%) withdrew from fibre group and 0% on lactulose. The GDG did not regard this level of missing data as significant.
Seven studies stated that they did not permit any concomitant medication that would change the GI motility (Bouhnik 2004; Chaussade 2003; Corazziari 1996; Kienzle-Horn 2006; Kienzle-Horn 2007; Quah 2006; Wulrow 2007). Five studies allowed the patients to have laxatives as relief medication: in two studies (Corazziari 1996; Corazziari 2000) there had to be five consecutive days without a bowel movement; in one study (Chaussade 2003) there had to be three consecutive days, after which the patients could have suppositories. In the other studies (Attar 1999; Medoff 2004) patients could use suppositories or microenemas for relief, apparently without restriction. In another study (Rouse 1991) 12/124 patients took other laxatives during the study and were considered to be protocol violators.
The risk of bias was assessed for each included study and no studies were excluded from the analysis (although the 20 week results for Corazziari 2000 were disregarded). The two studies in which laxatives could be taken apparently without restriction (Attar 1999; Medoff 2004) were regarded with caution.
RESULTS
I. Treatment for acute episodes of constipation
A. Laxatives versus placebo
Two studies (Kienzle-Horn 2006; Wulkow 2007) in 112 patients compared laxatives with placebo for the treatment of acute episodes of constipation. It was unclear if the patients had IBS. Stimulant laxatives 10mg bisacodyl (Kienzle-Horn 2006) or 7mg sodium picosulphate (Wulkow 2007) or placebo was given once-a-day for three days.
1. Global symptoms
Global symptoms (pain, bloating and bowel habit) were not reported.
2. Individual symptoms
a. Bowel habits
i. Number of patients with improvement in bowel habit assessed by investigators
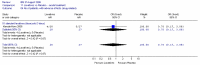
The investigators assessed the improvement in bowel habit, based on diary recordings of the patients. Overall, the relative risk was 1.34 (95%CI 1.02, 1.76) (Figure 1), i.e. statistically significant difference between laxative and placebo (p=0.04). This corresponded to a number needed to treat (NNT) of 6 (95%CI 3, 50) for a control group risk of 52 to 61%. There was no heterogeneity.

ii. Stool score - consistency
The consistency of stool was measured on the scale of 1 to 5, where 5=hard, 4=moderately hard, 3=well-formed, 2=soft, 1=liquid (Kienzle-Horn 2006) and on a 4 point scale where 4=hard, 3=well-formed, 2=pasty,1=liquid (Wulkow 2007). The Wulkow study reported the number of patients with soft and/or well formed stools. The relative risk was 1.51 (95%CI 1.06, 2.15) i.e. statistically significant difference between laxative and placebo (p=0.02) favouring laxative (Figure 1).
Kienzle-Horn (2006) reported baseline mean scores which were 5.0 for each group, so any decrease in score constituted an improvement. The study did not report the standard deviation for the placebo group, but gave the difference in change score between the two groups and the 95%CI (Figure 2). This was −1.4 (95%CI −2.0, −0.76), for a placebo group score of 4.2, i.e. a statistically significant difference between groups, such that the bisacodyl group had a value between soft and well-formed.


II. Laxatives for maintenance treatment
A. Laxatives versus Placebo
There were two studies included in the analysis that compared laxatives with placebo in patients with constipation (Corazziari 1996 and Corazziari 2000). Both studies gave the patients an isosmotic PEG electrolyte balanced solution (PMF-100) containing 14.6g PEG 4000, twice a day. However, in Corazziari (2000), all patients received 4 weeks of PMF-100 initially, with responders (more than 3 bowel movements per week) then randomised to PEG or placebo for a further 20 weeks. Thus the populations were different in the two trials, and Corazziari (2000) was regarded as an investigation of the effects of stopping the laxative. Their results are therefore reported separately. Both studies had patients who were outpatients in secondary care. The GDG considered it likely that both studies had at least some patients with IBS.
In both trials, patients were allowed other laxatives when they had no bowel movements for at least 5 consecutive days, and they were allowed to adjust the intervention dose downwards (but not upwards above 2 sachets per day).
Where outcomes were measured at different times during the study, we took the end-study results unless there were significant numbers of withdrawals or problems with compliance. Therefore, for the Corazziari (2000) study we took the values at eight weeks (i.e. from the start of randomisation).
1. Global symptoms
Neither study reported global symptoms.
2. Number of patients using additional laxatives / not using additional laxatives as rescue medication
The GDG considered this to be an important outcome for this review and gave it the status of primary outcome measure. We gave both the number of patients using additional laxatives (as reported in the papers), and the number not using additional laxatives (calculated).
The comparison of PEG and placebo over 8 weeks in 48 patients (Corazziari 1996) showed a statistically significant decrease in the number of patients using other laxatives as rescue medication; Figure 5; RR 0.33 (95%CI 0.12, 0.90), although the confidence interval was wide. This corresponded to a number needed to treat of 4 (95%CI 2, 15) for a placebo group risk of 48%.

Figure 5
Number of patients taking rescue laxatives.
For the outcome measure, the number of patients not using rescue medication was calculated for Corazziari (1996) (Figure 6). There was a statistically significant difference between PEG and placebo, favouring the former; RR 1.61 (95%CI 1.05, 2.47), which gave an NNT of 4 (95%CI 2, 15) for a control group risk of 52%.
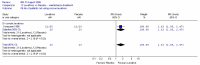
Figure 6
Number of patients not taking rescue laxatives.
Corazziari (2000) (withdrawal of laxative following four weeks PEG electrolyte solution, in responders) reported that the use of other oral laxatives, rectal evacuants, suppositories and enemas was more frequent in the placebo group compared to the PEG group. At eight weeks the difference in number of other laxatives used per four weeks was statistically significant (Figure 6), but the confidence interval was fairly wide.
At the same time, statistically significantly more sachets of the intervention were used in the placebo group (even though the more severely constipated patients dropped out), compared with the PEG group (Figure 7).

In the PEG group, the authors reported that the use of other laxatives progressively decreased in the PEG group but increased in the placebo group.
3. Individual symptoms
a. Pain
There was no significant difference between PEG and placebo for this outcome (Corazziari 1996) in 48 patients. The confidence intervals were fairly wide so there was some uncertainty over the results for the difference between groups in the number of patients with abdominal pain (Figure 8).

Corazziari (2000) (withdrawal of laxative following 4 weeks PEG electrolyte solution, in responders) reported that the abdominal pain score progressively decreased in the PEG group and increased in the placebo group. No data were given.
b. Bloating
The comparison of PEG and placebo (Corazziari 1996) in 48 patients showed no statistically significant difference between groups in the number of patients with bloating (Figure 9).
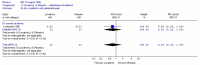
Corazziari (2000) (withdrawal of laxative following 4 weeks PEG electrolyte solution, in responders) reported that bloating was less severe in the PEG group compared to the placebo group throughout the study. At 8 weeks the difference was statistically significant: p<0.001. No other statistics were given.
c. Bowel habits
i. Stool frequency
The comparison of PEG and placebo over 8 weeks in 48 patients (Corazziari 1996) showed a statistically significant increase in stool frequency per week (figure 10) of 2.00 (95%CI 0.89, 3.11), for a placebo group value of 2.8 stools per week.

Corazziari (2000) (withdrawal of laxative following 4 weeks PEG electrolyte solution, in responders) found a statistically significant increase in stool frequency per week for the PEG group compared to the placebo group throughout the study. At 8 weeks (Figure 11) the difference was 3.13 (95%CI 1.35, 4.91) for a placebo group value of 4.39 stools per week.

ii. Improvement in bowel habit
Corazziari (2000) (withdrawal of laxative following 4 weeks PEG electrolyte solution, in responders) found a statistically significantly greater number of patients with complete remission of constipation symptoms (more than three bowel movements per week, no use of other laxatives, no straining at defecation, no feeling of incomplete evacuation, no hard/pellety stools) for the PEG group compared to the placebo group throughout the study. At 8 weeks (Figure 12) the RR was 3.95 (95%CI 1.86, 8.42); this corresponds to an NNT of 2 (95%CI 2, 3) for a control group rate of 18%.


Figure 13
iii. Number of patients withdrawing from study
Corazziari (2000) (withdrawal of laxative following 4 weeks PEG electrolyte solution, in responders) reported statistically significantly more patients withdrew from the study by 20 weeks because of non-response to treatment in the placebo group compared to the PEG group. At the end of the study the RR was 0.13 (95%CI 0.03, 0.53), i.e. statistically significantly in favour of the PEG group, although the confidence interval was very wide. This corresponded to an NNT of 3 (95%CI 2, 5) for a placebo group rate of 46%.

Figure 14
4. Adverse effects
In the comparison of PEG and placebo over 8 weeks in 48 patients (Corazziari 1996), there was too much uncertainty to determine if there was a difference in the number of patients reporting anorexia, headache or asthenia (Figure 15).
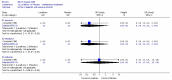
Corazziari (2000) reported that there were no significant differences between groups in the incidence of adverse effects.
B. Osmotic Laxative type 1 versus Osmotic laxative type 2
Three studies compared different types of osmotic laxatives: two (Attar 1999; Bouhnik 2004) compared lactulose with PEG, and one compared different types of PEG: PEG 3350 plus electrolytes versus PEG 4000 without electrolytes (Chaussade 2003). We noted that the Bouhnik (2004) was sponsored by Solvay Pharmaceuticals, manufacturers of lactulose, and Chaussade (2003) was supported by a grant from by Hoffmann La Roche, the manufacturers of PEG 3350. The Attar (1999) study was in secondary care (of which 31% were in geriatric institutions), and the Chaussade (2003) and Bouhnik (2004) studies were in primary care. The GDG thought it likely that all of these studies had some patients with IBS.
B1. Lactulose versus PEG
Attar (1999) compared PEG 3350 plus electrolytes (Movicol) versus lactulose, and Bouhnik (2004) compared PEG 4000 plus electrolytes (Forlax) versus lactulose. Both studies had a duration of four weeks. The doses of PEG differed in the two studies: the patients in Attar (1999) started with 26.24 g (2 sachets) for the first two weeks, but could change to 1 or 3 sachets for the second two weeks. Patients in Bouhnik (2004) started at a dose of 20g (2 sachets) for the first week and then this could be varied to 10 or 30g. The lactulose dose in both studies was 20g which could also be varied as above.
In Attar (1999) patients could take suppositories or microenemas for relief of constipation, apparently without restriction. However, in Bouhnik (2004) patients were asked to stop enema/suppositories 48 hours before the first stool collection.
1. Global outcomes
a. Global improvement in symptoms score
One study (Attar 1999) in 99 patients recorded a global improvement score at four weeks on a VAS of 0 to 10 (0=no change, 10=excellent); comprising pain, bloating and bowel habit. The global improvement score was statistically significantly in favour of PEG electrolyte; mean difference 2.20 (95%CI 1.05, 3.35) for a control group value of 5.20.

Figure 16
2. Use of microenemas as rescue medication
Attar (1999) also reported the number of patients using microenemas as rescue medication after four weeks, and we also calculated the number of patients not using rescue medication. The study found that statistically significantly more patients used microenemas in the lactulose group than in the PEG group; RR 0.48 (95%CI 0.25, 0.95), which corresponded to an NNT of 6 (95%CI 3, 50) for a lactulose group risk of 35% (Figure 17a). The confidence interval was fairly wide.

Figure 17a
Number of patients using microenemas.
There was a statistically significant difference, favouring PEG, for the number of patients not using microenemas; RR 1.27 (95%CI 1.02, 1.59). This corresponded to an NNT of 6 (95%CI 3, 50) for a lactulose risk of 65% (Figure 17b).
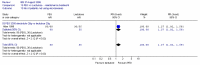
Figure 17b
Number of patients not using microenemas.
3. Number of sachets of intervention used
In Attar (1999) the number of sachets of laxative used over four weeks was statistically significantly lower for the PEG group (Figure 18), but there was no difference between groups in Bouhnik (2004). This led to significant heterogeneity between studies (I2=83%, p=0.02). It is unclear if this was an effect of dose; type of PEG; use of other laxatives, or; any other reason.

4. Individual symptoms
a. Pain
Both studies reported the number of patients with abdominal pain at four weeks. Meta-analysis of 180 patients gave a wide confidence interval.

Figure 19
One study (Attar 1999) recorded pain on a scale of 0 to 3 (severe). The difference was not statistically significant.

Figure 20
b. Bloating
Both studies reported the number of patients with bloating at four weeks. Meta-analysis of 180 patients gave a fairly wide confidence interval and some heterogeneity (I2=50%; p=0.16). This difference may be an effect of dose or type of PEG. It is also noted that Bouhnik (2004) was sponsored by the manufacturers of lactulose and Attar (1999) allowed the patients to use other laxatives ad libitum.

Figure 21
c. Bowel habits
i. Stool frequency
Both studies reported the stool frequency per day at four weeks. Meta-analysis of 180 patients gave a statistically significant difference of 0.27 (95%CI 0.09, 0.45) stools/day, favouring PEG, but there was some heterogeneity (I2=50%; p=0.16).
5. Adverse effects
Both studies reported the number of patients with adverse effects at four weeks. Meta-analysis of 180 patients gave a wide confidence interval and no heterogeneity (Figure 22). One study (Attar 1999) reported the number of patients with liquid stools, but this also had a wide confidence interval (Figure 23).

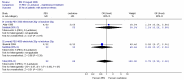

Figure 24
B2. Comparison of different PEG laxatives
One study (Chaussade 2003) compared two doses of each of two types of PEG solution for a duration of four weeks. The PEG species were PEG 4000 (Forlax) without electrolytes and PEG 3350 plus electrolytes. Doses used were the maximum and standard recommended by the manufacturers.
1. Global improvement of symptoms
The study measured the patients’ global impression of efficacy on a VAS, but the results are not reported. The authors state that there was no significant difference between groups.
2. Individual symptoms
a. Abdominal pain
Pain scores at four weeks were recorded on a scale of 1 (none) to 4 (considerable). There was no significant difference between the two types of PEG for this outcome at either dose, and there was no heterogeneity (I2=0%).

Figure 25
b. Bloating
Bloating scores at four weeks were recorded on a scale of 1 (none) to 4 (considerable). There was no significant difference between the two types of PEG for this outcome at either dose, and no heterogeneity (I2=0%).

Figure 26
c. Bowel habits
i. Stool frequency per week
There was no significant difference between the two types of PEG for this outcome at four weeks at either dose, and no heterogeneity (I2=0%).

Figure 27
ii. Stool consistency
Consistency of stools at four weeks were recorded on a scale of 1 (liquid) to 6 (very hard). Meta-analysis of the two comparisons revealed heterogeneity (I2=65%, p=0.09). For the standard dose, there was a statistically significant difference between the two PEG solutions, favouring PEG 4000; mean difference 0.30 (95%CI 0.01, 0.59). This was a fairly small change. There was no significant difference for the maximum dose.

Figure 28
iii. Number of patients with normal stools
Meta-analysis showed no significant difference at four weeks between types of PEG.
3. Quality of life
The study measured quality of life on a 100mm VAS at four weeks. Meta-analysis showed no significant difference between types of PEG and no heterogeneity (I2=0%, p=0.93).

Figure 30
4. Adverse effects
For overall adverse effects at four weeks, the majority of which were gastrointestinal, meta-analysis showed no significant difference between types of PEG and no heterogeneity (I2=0%, p=0.55).

Figure 31
For the specific adverse effect of diarrhoea, there was no significant difference between types of PEG and no heterogeneity (I2=0%, p=0.71).

Figure 32
B3. Comparison of different doses of PEG laxatives
One study (Chaussade 2003) compared two doses of each of two types of PEG solution for a duration of four weeks. The PEG species were PEG 4000 (Forlax) without electrolytes and PEG 3350 plus electrolytes. Doses used were the maximum and standard recommended by the manufacturers.
1. Global improvement of symptoms
The study measured the patients’ global impression of efficacy at four weeks on a VAS, but the results are not reported. The authors state that there was no significant difference between groups.
2. Individual symptoms
a. Abdominal pain
Pain scores at four weeks were recorded on a scale of 1 (none) to 4 (considerable). Meta-analysis showed no significant difference between the two doses for this outcome, and no heterogeneity (I2=0%).

Figure 33
b. Bloating
Bloating scores at four weeks were recorded on a scale of 1 (none) to 4 (considerable). There was no significant difference between the two doses for this outcome, and no heterogeneity (I2=0%).

Figure 34
c. Bowel habits
i. Stool frequency
There was no significant difference between the two doses for this outcome at four weeks, and no heterogeneity (I2=0%).

Figure 35
ii. Stool consistency
Consistency of stools at four weeks was recorded on a scale of 1 (liquid) to 6 (very hard). Meta-analysis of the two comparisons revealed some heterogeneity (I2=65%, p=0.09). For the PEG 3350 electrolyte dose, there was a statistically significant difference between the two doses, favouring the maximum dose; mean difference 0.60 (95%CI 0.29, 0.91). There was no significant difference for the PEG 4000.

Figure 36
iii. Number of patients with normal stools
Meta-analysis of the two comparisons reveals statistically significantly more patients with normal stools at four weeks for the standard dose groups. The RR was 1.68 (95%CI 1.14, 2.48), which corresponded to an NNH of 7 (95%CI 4, 25) for the higher dose rate of 19% or 25%.

Figure 37
3. Quality of life
The study measured quality of life at four weeks on a 100mm VAS. Meta-analysis showed no significant difference between doses and no heterogeneity (I2=0%, p=0.93).

Figure 38
4. Adverse effects
For overall adverse effects at four weeks, the majority of which were gastrointestinal, meta-analysis showed no significant difference between doses and no heterogeneity (I2=0%, p=0.55).

Figure 39
For the specific adverse effect of diarrhoea, there was a statistically significant difference, favouring the standard dose, and no heterogeneity (I2=0%, p=0.68). The RR was 0.41 (95%CI 0.24, 0.70); this corresponded to an NNT of 6 (95%CI 4, 13) for the higher dose rate of 30%.
B4. Stimulant Laxative Type 1 versus Stimulant Laxative Type 2
One study compared two stimulant laxatives, bisacodyl versus sodium picosulphate (Kienzle-Horn 2007). Patients were treated daily for 4 weeks with 5 to10mg of either bisacodyl or sodium picosulphate. The primary outcome was the change in bowel habit recorded as the mean number of bowel movements per day and stool consistency measured on a 5 point scale where 5=hard, 4= moderately hard,3= well formed,2=soft, 1=liquid. Secondary outcomes included straining scored on a 4 point scale with 4 = severe and 0=absent. There was no statistically significant difference between the two laxatives for the number of bowel movements per day, WMD: −0,05 (95%CI −0.18, 0.08), and similarly for the stool consistency and straining score. Both were equally effective in treating constipation.

Figure 41
C. Laxative versus fibre
Three studies compared a laxative with fibre: two compared an osmotic laxative (lactulose) with fibre (ispaghula), (Quah 2006; Rouse 1991) and one compared usual laxatives (mainly lactulose) with fibre (ispaghula) (Dettmar 1998). Quah (2006) had a crossover design, with a washout period of 1 week, and this study was treated separately. Quah (2006) and Rouse (1991) compared respectively: 20 ml lactulose with 3.5g ispaghula husk, and 30 ml lactulose with 7g ispaghula husk. Dettmar (1998) did not record the dose of lactulose (other laxatives), but 7g ispaghula husk was given. Dettmar (1998) also reported results for the lactulose subgroup of ‘other laxatives’. Rouse (1991) and Dettmar (1998) were in primary care and Quah (2006) in secondary care. The authors of Dettmar (1998) were from Reckitt and Colman, manufacturers of Fybogel. The GDG considered it unlikely that the patients in Dettmar (1998) and Rouse (1991) had IBS. It was unclear if the patients in Quah (2006) had IBS.
1. Global symptoms
Two studies reported an outcome of global symptoms (Rouse 1991; Dettmar 1998).
a. Global improvement of symptoms
One study (Rouse 1991) in 112 patients showed little difference in global improvement of symptoms at four weeks between patients given 30ml lactulose and 7g ispaghula husk (figure 40).


Figure 42
a. Global effectiveness
Dettmar (1998), in 315 patients, asked the patients to rate the effectiveness at four weeks of treatment with ispaghula 7g and other laxatives, mainly lactulose. Statistically significantly more patients given ispaghula rated the effectiveness as excellent, good or satisfactory. RR (all other laxatives versus fibre) was 0.87 (95%CI 0.80, 0.96) and for the subgroup with lactulose the RR was 0.90 (95%CI 0.85, 0.96). It was noted that authors of Dettmar (1998) were from the manufacturers of ispaghula (Reckitt and Colman).

Figure 43
When these two studies were combined in a meta-analysis, the RR was 0.92 (0.85, 1.00) and there was a significant heterogeneity I2 = 73.5%, p=0.05. It is unclear what caused this, but the overall effect was small.

Figure 44
2. Individual symptoms
a. Pain
Two studies (Rouse 1991; Quah 2006) in 93 and 78 patients respectively, recorded the number of patients with abdominal pain at four weeks. We did not combine these studies because one was a crossover study and the other parallel. We did not draw conclusions for the crossover study because the confidence interval was too wide; there was also only 1 week washout for this study. The confidence interval was fairly wide for Rouse (1991), but there was no significant difference between lactulose and ispaghula.
b. Bloating
Two studies recorded the number of patients with bloating at four weeks: Quah (2006) in 76 patients, and; Dettmar (1998) in 394 patients. There was little difference in the numbers with bloating, although the confidence intervals were fairly wide.

Figure 46
c. Bowel habits
i. Improvement in bowel score
One crossover study (Quah 2006) with 78 patients recorded improvement in bowel score at four weeks compared with baseline on a scale of 0 (no effect) to 10 (excellent) (Figure 45). There was a statistically significantly greater improvement with lactulose compared to ispaghula; mean difference 1.40 (95%CI 0.19, 2.61). It was noted that this crossover study had a washout period of only 1 week, so the results were treated with caution.


Figure 47
ii. Stool frequency
One study reported the stool frequency at four weeks (Quah 2006). There was a non-significant difference between lactulose and ispaghula, favouring the former; WMD 1.80 (95%CI −0.12, 3.72). It was noted that this crossover study had a washout period of only 1 week, so the results were treated with caution.
iii. Stool consistency
One study reported the stool consistency at four weeks on a scale of 0 (no bowel movement) to 3 (comfortable and solid) to 5 (loose) (Quah 2006). There was a borderline significant difference of 0.50 (95%CI 0.00, 1.00; p=0.05) between lactulose and ispaghula, favouring the former. However, since the normal rating is 3 and the fibre group is closer to this value (2.9) it could be argued that fibre is more favourable. It was noted that this crossover study had a washout period of only one week, so the results were treated with caution.

Figure 49
3. Adverse effects
Two studies reported the number of patients with adverse effects at four weeks. In all cases there were wide confidence intervals (Figure 48).


Figure 50
4. Patient preference
The crossover study, Quah (2006), recorded patient preference at four weeks between lactulose and ispaghula. Statistically significantly more patients preferred lactulose; RR 1.71 (95%CI 1.05, 2.79). This gave an NNT of 4 (95%CI 3, 25). It was noted that this crossover study had a washout period of only one week, so the results were treated with caution.

Figure 51
Adverse Effects
An adverse effects review has been carried out and is reported in section 8.5.1. The review included six RCTs (Quah 2006; Ferguson and Attar 1999; Bouhnik 2004; Corazziari 1996; Corazziari 2000; Chaussade 2003), and their results are reported in this effectiveness review. The RCTs were primarily aimed at assessing and reporting on the efficacy of the drug treatments. Evaluation of safety and reporting of adverse effects data was often cursory or nonexistent. Even in instances where the methods sections had explicitly stated the intention of monitoring for adverse effects, trial reports did not follow a structured format (e.g. by WHO system organ class) of reporting adverse effects. The interventions and comparators were extremely varied, as was the reporting of adverse effects.
One non-randomised study in the US reported a series of adverse effects of laxatives, but did not distinguish laxative class, and used doses higher than in the UK.
Many of the adverse outcomes of interest are very similar to the symptoms of the IBS itself. For instance, laxatives are associated with flatulence, cramps and abdominal pain – all of which are commonly seen in untreated IBS patients and also form part of the efficacy assessment. It is not always possible to determine whether deterioration in these symptoms is due to lack of efficacy, or the natural history of the disease, or the adverse effect of the drug. Generally, though, the RCT data on lactulose was consistent with the findings of the non-randomised data with regards to increased risk of abdominal symptoms. The GDG’s clinical experience of lactulose was that it caused bloating and other side effects.
ECONOMIC LITERATURE FOR LAXATIVES
One relevant health economic analysis was identified on the cost-effectiveness of laxatives in the treatment of IBS. Christie (2002) was a model based economic evaluation from a UK perspective which used efficacy data from a secondary care trial comparing two laxatives conducted in Scotland and France. The population included in the trial was patients with idiopathic constipation of greater than three months duration and the population included some elderly patients living in institutions. Whilst data from elderly residential patients is not directly relevant to the IBS population, this paper was included as indirect evidence for the IBS population.
This study aimed to assess the economic impact of using low dose polyethylene glycol 3350 plus electrolytes (PEG+E) compared to lactulose in the treatment of idiopathic constipation using a decision analytic model. The economic analysis was carried out from an NHS perspective. As discussed earlier, this study was considered to be indirect evidence as the patient population was not restricted to patients with IBS but may have included some patients with IBS-C. The effectiveness inputs used in the model were obtained from a randomised controlled trial conducted in primary care. In this trial patients were randomised to treatment with either PEG+E or lactulose for one month. After this initial comparator controlled phase, patients aged over 65 continued on their allocated treatment for two months but those aged under 65 received lactulose for a further 2 months regardless of their initial allocation. The model considered the probability of various clinical outcomes over 2 weekly intervals for a 3 month period. The outcomes considered by the model were; successful treatment, discontinuation of treatment due to an adverse event, switching laxatives due to an adverse event, discontinuation of treatment due to lack of efficacy, switching laxatives due to lack of efficacy, not complying with either treatment and discontinuing treatment, not complying with either treatment and switching to another laxative. Resource use estimates were provided by a panel of experts.
PEG+E had a higher probability of achieving successful treatment at 3 months (53% versus 24%) but a higher acquisition cost (£25.42 versus £10.05 over 3 months). This was offset by a reduced number of GP appointments (2.9 visits versus 4.4 visits), resulting in an overall lower cost in patients initially treated with PEG+E (£85 versus £96). The sensitivity analyses showed that the overall costs were particularly sensitive to changes in the efficacy of first-line treatment with either treatment, the mean daily dose for PEG+E, the probability of senna being co-prescribed with lactulose, the probability of discontinuing treatment with lactulose and the number of GP appointments. Given that the costs were sensitive to dose it is important that this study is considered along-side evidence on the effective dose in patients with IBS. The model assumed that co-prescription of senna is more frequent in patients not experiencing successful resolution of symptoms following lactulose treatment (13%) than following PEG+E treatment (2%). In the trial patients were not allowed to take additional laxatives, so the effectiveness of adding senna to lactulose in this way would not be captured in the model but the cost of co-prescribing senna has been included in the model. Assuming no senna use in the lactulose arm reduced the cost to £87 which suggests that the cost-effectiveness is sensitive to the accuracy of this assumption.
The study was a partial economic evaluation as it did not assess the incremental cost of any benefit achieved in the form of a cost-effectiveness ratio. This may be appropriate given that the intervention was more effective than the comparator. However, the effectiveness was only measured in terms of the probability of successful treatment rather than overall health impact. This may be misleading if adverse events have a higher impact on health than successful treatment. Adverse events were included in the analysis but from a cost perspective only. The evidence provided by this study was not directly relevant to the guideline as it considered a patient population that isn’t fully representative of the population considered by this guideline. No potential areas of significant bias were identified, but the sensitivity analysis demonstrated that the magnitude of cost-saving estimated by the model was variable under the parameter ranges considered. Modelled direct health care costs were lower in the PEG+E arm despite a higher acquisition cost. As this study did not provide an estimate of the cost per QALY for PEG+E compared to lactulose, and did not consider the cost-effectiveness of either intervention compared to no laxative treatment, it was not particularly useful in determining whether recommending PEG+E or lactulose would result in the efficient use of NHS resources.
COST-EFFECTIVENESS ANALYSIS FOR LAXATIVES
This section describes the health economic analysis undertaken to inform recommendations on the use of laxatives as a long-term maintenance therapy in IBS. The general methods used in the economic analysis for all management interventions are described in detail in Chapter 5 and the model inputs and assumptions relevant to this particular intervention are described below.
The general approach was the same as for other maintenance therapies except:
- None of the trials provided an estimate of the relative risk of an improvement in global symptom score, which was the favoured outcome for determining a successful response to treatment for the economic model. An improvement in bowel habit was considered as a possible alternative definition for response, but this was also not available for any of the long-term maintenance studies. In the absence of this, a successful response was defined as no use of other laxatives.
- For sodium picosulfate and bisacodyl there was evidence for their effectiveness compared to placebo for short term use (3 days) but there was no evidence on their effectiveness compared to placebo for long-term use. In the absence of evidence on the effectiveness of long-term maintenance use, we have applied the effectiveness from the short-term trials and assumed that it would persist in the long-term. This is an extreme extrapolation beyond the available trial duration and should be considered with caution.
- PEG, sodium picosulfate and bisacodyl were included in the economic model as potential laxative treatments. We assumed that PEG is used first line as this was the only intervention with evidence of clinical effectiveness in long-term maintenance use. Sodium picosulfate and bisacodyl are assumed to be used second line in patients who do not respond to PEG.
- Lactulose was less effective than PEG 3350 (with electrolytes) (Attar 1999). GDG consensus was that people with IBS should be actively discouraged from taking Lactulose as it promotes gaseous bloating which can exacerbate IBS symptoms. It was therefore excluded from the cost-effectiveness analysis.
- The studies included in the clinical effectiveness review did not stratify results by IBS subtype, but all the studies were carried out in patients with chronic constipation. Therefore, the cost-effectiveness is estimated for patients with IBS-C. The applicability of these results to people with IBS-A was considered by the GDG as they may have intermittent periods of chronic constipation.
Modelled response rates
In the basecase scenario the response rate of 45% in the no treatment arm is taken from the Mearin (2004) cohort study. This represents the group of patients whose symptoms improve without any specific intervention. The RR of response for PEG versus placebo is 1.61; therefore the response rate in the PEG arm is 72% (=45% x 1.61), giving an absolute difference in response between the intervention and no treatment arms of 27% (=72%–45%) during the first month for PEG. The RR of bisacodyl and sodium picosulfate is 1.34 compared to placebo, so if these interventions are used first line then we would expect an absolute difference in response between intervention and no treatment of 15% (=1.34*45%–45%). The first line use of bisacodyl and sodium picosulfate has not been modelled due to a lack of longer-term data on their effectiveness compared to placebo.
In the basecase scenario the response rate for the subsequent interventions is assumed to be equal to the response rate to the first intervention. If bisacodyl and sodium picosulfate are used second line in patients who do not respond to PEG, we would expect an additional 4.2% (=15% x 28%) of the original cohort to respond to the second laxative, and an additional 3.6% (=15% x 24%) to respond to the third laxative, giving an overall response rate of 80% for laxatives. The response rate over time for the basecase is given in Figure 52. It is assumed that bisacodyl is tried first after a failure to respond to PEG as it has a lower cost than sodium picosulfate.
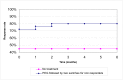
Figure 52
Modelled response rates for laxatives (PEG followed by two switches to other laxatives for non responders) and no treatment.
We have also considered an alternative scenario in which no patient in the comparator arm achieves an improvement in symptoms, but the absolute gain in response rates is maintained from the basecase (e.g. for first line PEG, we modelled a zero response to no treatment but a 27% response to PEG for this scenario).
Table 1Intervention specific parameters – laxatives
| Description | Value | Evidence | |
|---|---|---|---|
| RR of response for PEG vs placebo | 1.61 | Meta-analysis of RCT evidence for no use of other laxatives | |
| RR of response for bisacodyl and sodium picosulfate compared to placebo | 1.34 | Meta-analysis of RCT evidence for improvement in bowel habit | |
| Maximum number of switches considered | 2 | Limited by number of effective interventions | |
| Drug costs | |||
| Intervention | Dose per day | Cost per month* (assuming lowest cost preparation) | |
| PEG | 23g (equiv to 1.8 sachets of Movicol or 2.3 sachets of Idrolax) | £12.54 | |
| Bisacodyl | 10mg | £1.43 | |
| Sodium picosulfate | 7mg | £3.94 | |
- *
British National Formulary (Joint Formulary Committee 2007)
Table 2 gives the incremental cost-effectiveness for several laxative treatment pathways in order of the benefits they achieve. It shows that whilst PEG provides additional benefit for a cost per QALY of £7,779, compared to no treatment, further benefit can be achieved by allowing non responders to PEG to switch to bisacodyl and if that is not effective to switch to sodium picosulfate. Each of these additional switches for non responders has a low cost per QALY compared to no further treatment for non-responders (£4,488 and £6,561 respectively). However, it should be noted that the cost-effectiveness of these second line laxatives is based on clinical effectiveness evidence for bisacodyl and sodium picosulfate from short term trials lasting only 3 days.
Table 2
Incremental cost-effectiveness of allowing subsequent switches in laxative therapy.
These results are an estimate of the cost-effectiveness over the first 6 months after the initiation of laxative therapy. The cost per QALY for continuing laxative therapy beyond 6 months is lower than the cost per QALY during the initial 6 months provided that treatment is reviewed every 6 months and discontinued in patients who no longer experience a therapeutic benefit, either due to lack of effectiveness or a change in their symptom profile.
The probabilistic sensitivity analysis provides an estimate of the uncertainty in the cost per QALY estimate due to uncertainty in the efficacy estimate, the probability of response in the no treatment arm and the utility gain. The CEAC in Figure 53 shows the uncertainty surrounding the cost-effectiveness of PEG, compared to no treatment and the incremental cost-effectiveness of allowing non-responders to switch to other laxatives. It shows that there is an 83% likelihood that the cost per QALY for PEG compared to no treatment is under £20K, suggesting that PEG provides health benefit at an acceptable cost in patients with IBS-C. Allowing non responders to PEG one treatment switch has an 81% probability of a being cost-effective when a £20K threshold is applied compared to no further treatment for non responders. Similarly, allowing a second treatment switch for non-responders has a 74% probability of being cost-effective.
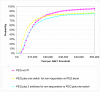
Figure 53
CEAC for PEG with up to two switches for non responders compared to no treatment (NT).
However, it should be noted that these estimates only consider the uncertainty in cost-effectiveness due to the accuracy of several input parameters and they do not reflect general uncertainty around the assumptions made in the model. The uncertainty from these assumptions was explored in the univariate sensitivity analysis.
Univariate sensitivity results for laxatives
The results of the univariate sensitivity analysis for PEG compared to no treatment are given in Table 3. Maintaining the 27% difference in response between the two arms but reducing the response rate in the no treatment arm from 45% to zero decreased the cost per QALY to £4,896. The use of higher cost formulations increased the cost per QALY to £9,980, whilst assuming that 50% of prescriptions were over the counter reduced the cost per QALY to £4,814.
Table 3
Sensitivity results for PEG compared to no treatment for 100 patients with IBS-C.
The cost per QALY is less favourable for patients who only use the medication on 25% of days as the upfront costs of initiating therapy and establishing response are constant despite lower benefit from less frequent use. PEG compared to no treatment has a cost per QALY of £13,325 when used on 25% of days.
We carried out a threshold analysis to determine whether laxative therapy would still be cost-effective for lower gains in health related quality of life. In the basecase it was assumed that patients who respond to therapy accumulate 0.071 QALYs more per annum than patients who do not respond. For comparison, a gain of 0.135 QALYs would represent a complete remission of IBS symptoms. If the QALY gain associated with a response to therapy was reduced to 0.027 QALYs, then the cost per QALY of providing PEG compared to no treatment would be above £20,000 per QALY. The methods used in this analysis vary from the methods used for other pharmacological interventions as we were unable to estimate the response rate in terms of an improvement in global symptoms from the trial data available. Instead we assumed that patients who did not use another laxative during the trial had responded to the trial intervention. This outcome was considered a less reliable indicator of whether there had been an overall improvement in HRQoL than an improvement in global symptoms. However, the threshold analysis shows that laxatives are cost-effective even if the utility gain associated with a therapeutic response is small.
We carried out a similar univariate sensitivity analysis on the incremental cost-effectiveness of allowing non responders to switch to an alternative laxative. The incremental cost per QALY for the first and second switches was increased to £8,784 and £12,624 when assuming that patients who demonstrate no response to PEG would be half as likely to respond to another laxative. The incremental cost per QALY for each subsequent treatment switch in non-responders was higher for patients using treatments intermittently with a cost per QALY of £8,468 for the first switch and a cost per QALY of £11,536 for the second switch in patients who use laxatives on only 50% of days. When laxatives are used on only 25% of days, the incremental cost per QALY estimates for the first and second switches are £16,428 and £21,485 respectively.
If a patient also takes another medication (an antispasmodic), then this medication can be reviewed at the same time, so it may be cost-effective to provide both therapies. For example, if laxatives are prescribed with the antispasmodic and both used on 25% of days then allowing up to 2 switches of both treatments was estimated to be cost-effective with a cost per QALY of £10,107 compared to no treatment a cost per QALY of £17,393 compared to 1 switch.
GDG DISCUSSION
Many of the studies included in the laxative review may be considered to be indirect evidence as the participants were defined as having simple constipation. However the GDG considered that many of these participants may have had IBS, but the studies did not use any IBS assessment criteria and the trials were designed to treat the symptoms of constipation. General consensus is that IBS is very different from simple constipation. People with IBS cannot cope with gas and some laxatives increase gas and exacerbate IBS symptoms, lactulose in particular. IBS patients should be actively discouraged from taking lactulose. The GDG also referred to best practice of titrating the dose of laxative to optimise symptoms, using the Bristol Stool Form Scale.
EVIDENCE STATEMENTS
For this review, the evidence was assessed using the GRADE process and tables are shown in Appendix F. The following evidence statements are derived from the GRADE tables.
- In studies for short-term symptom relief of constipation there is a moderate amount of good evidence to show a significant improvement in bowel habit for stimulant laxatives (bisacodyl and sodium picosulphate) compared to placebo. The study population included patients with IBS.
- There were no studies identified that used global improvement of symptoms as an outcome in longer-term maintenance treatment of constipation for PEG.
- In studies for longer-term maintenance treatment with PEG versus placebo in people with constipation (including people with IBS) there is a:
- Limited amount of good evidence that those taking PEG required significantly less rescue medication than those taking placebo.
- Limited amount of good evidence showing no significant difference in pain.
- Limited amount of good evidence showing significant reduction in bloating
- Moderate amount of good evidence showing a large significant improvement in bowel habit.
- There are no trials of longer term treatment that compared:
- Lactulose versus placebo
- Bisacodyl versus placebo
- Sodium picosulphate versus placebo.
- In studies for longer-term maintenance of PEG versus lactulose in people with constipation (including people with IBS) there is a:
- Fair amount of evidence showing significant improvement in global symptoms
- Moderate amount of good evidence that those taking PEG required significantly less rescue medication than those taking lactulose
- Moderate amount of good evidence showing a significant improvement in stool frequency.
- In studies for longer-term maintenance of bisacodyl versus sodium picosulphate in patients with constipation (including participants with IBS) there is a moderate amount of good evidence that there is no significant difference in stool frequency.
- In studies for longer term maintenance of PEG + Electrolyte versus PEG-Electrolyte in people with constipation (including participants with IBS) there is a:
- Moderate amount of good evidence to show that both are equally effective with no significant difference in pain, bloating, stool frequency, the number of people with normal stools, quality of life and adverse effects.
- In studies for longer-term maintenance treatment with standard and maximum dose PEG in people with constipation (including people with IBS) there is a:
- Moderate amount of good evidence to show that both are equally effective with no significant difference in pain, bloating, quality of life and adverse effects
- Moderate amount of good evidence showing a significant increase in the number of people with normal stools (standard dose)
- Fair amount of good evidence showing a significant increase in the incidence of people with diarrhoea (maximum dose).
ADVERSE EFFECTS EVIDENCE STATEMENTS
- There is limited evidence that laxatives are significantly associated with GI adverse effects (Abdominal cramps, abdominal discomfort, bloating, diarrhoea, abdominal pain, nausea).
- There is consistent evidence that lactulose increases the risk of abdominal symptoms in people with IBS.
- There is moderate evidence that low dose PEG is associated with fewer adverse effects compared to high dose PEG.
HEALTH ECONOMIC EVIDENCE STATEMENT
Evidence from a published model based economic evaluation comparing PEG with lactulose showed that PEG dominates lactulose by achieving a higher response to treatment rate at lower overall cost. The study was a partial economic evaluation as it did not assess the overall impact on health or provide the incremental cost of any benefit achieved in the form of a cost-effectiveness ratio. It is also considered to be indirect evidence as the population was not fully representative of the IBS population.
Evidence from a decision analytic model showed that laxatives (polyethylene glycol (PEG), bisacodyl and sodium picosulfate) are cost-effective for long-term maintenance use in individuals with IBS. The cost-effectiveness estimate is based on a clinical pathway in which response is assessed after one month and non-responders are switched to an alternative laxative with PEG used first line followed by bisacodyl and then sodium picosulfate. The cost-effectiveness analysis assumes that treatment is reviewed every 6 months to establish whether it is still relevant to the individual’s symptom profile.
EVIDENCE TO RECOMMENDATIONS
The evidence from the review suggests that laxatives are clinically and cost effective in the management of constipation. However the GDG clinical opinion is that IBS is more complex than simple constipation. Some laxatives exacerbate IBS symptoms and should therefore be avoided by people with IBS. The GDG recommended the continuation of current best practice of titrating the dose of laxative to optimise symptoms, based on the Bristol Stool Form Scale.
RECOMMENDATION
Laxatives should be considered for the treatment of constipation in people with IBS, but people should be discouraged from taking lactulose.
RECOMMENDATION
People with IBS should be advised how to adjust their doses of laxative or antimotility agent according to the clinical response. The dose should be titrated according to stool consistency, with the aim of achieving a soft, well-formed stool (corresponding to Bristol Stool Form Scale type 4).
8.2. Antimotility agents
SELECTION CRITERIA
The selection criteria described in the general methodology section were used, but some were specific to the antimotility agents review and are reported below.
Types of participants
For this review, participants were required to have IBS and not to have inflammatory bowel disease or diarrhoea subsequent to surgery. This inclusion criterion was adhered to for the longer term maintenance studies, but, for short term relief of symptoms investigations, there were insufficient data for IBS patients. Therefore, for this section of the review only, the GDG extended the population, post-hoc, to include studies in patients with acute diarrhoea of any cause (including those with diarrhoea caused by infection or virus). Such studies were regarded as indirect as far as the population was concerned.
Types of studies
The GDG decided that the washout period for this review should be at least one week. Trials with shorter washout periods were not included in the analysis.
Types of intervention
Studies included the following interventions:
- Codeine phosphate
- Co-phenotrope (diphenoxylate and atropine mixture; Trade name: Lomotil®)
- Loperamide
- Single drug: loperamide hydrochloride (Trade names: Norimode®, Imodium®)
- Compound preparation: loperamide hydrochloride and simeticone (Trade name: Imodium® Plus)
- Morphine
- Kaolin and Morphine mixture BP
- Morphine preparations on sale to the public.
The following comparisons were included:
- Antimotility agent versus placebo (or nothing)
- Antimotility agent type 1 versus type 2
- Antimotility agent dose 1 versus dose 2
- Antimotility agent + another intervention versus the other intervention alone
- Antimotility agent delivery mode 1 versus delivery mode 2
- Duration of treatment 1 versus duration 2.
NB: In spite of the large placebo effect associated with IBS, comparisons with no treatment are included.
The antimotility agents review was concerned with both longer term maintenance treatment and short-term symptom relief.
For maintenance studies, the GDG had decided that there should be a minimum duration of treatment of four weeks, but on further reflection agreed to include studies of two weeks or more. Studies of shorter durations were excluded. Short-term/symptom relief studies had duration of less than one week.
Subgroup analyses
We planned to carry out subgroup analyses by type of antimotility agent, dose, mode of delivery (modified release/conventional) and duration of intervention.
SEARCH STRATEGY FOR IDENTIFICATION OF STUDIES
Searches were performed on the following core databases: MEDLINE; EMBASE; CINAHL, and; The Cochrane Library (1966 to current day with guidance from the GDG). Additional databases were not searched for this review. The search strategies are listed in Appendix B.
The search strategy identified 2869 possible studies. The titles and abstracts of these studies were assessed. Forty were identified to be potentially relevant to the review and these papers were retrieved in full. The reference lists for each of the retrieved studies were inspected for further potential papers but none were identified. The 18 excluded studies are listed in Appendix E, along with reasons for exclusion.
DESCRIPTION OF STUDIES INCLUDED IN THE REVIEW
There were 22 included studies, six of which had at least some patients with IBS and 16 were in an indirect population (Allison 1988; Amery 1975; Barbezat 1979; Cann 1984; Corbett 1980; Cornett 1977; Dettmer 1994; Dom 1974; Dreverman 1995; Efskind 1995; Ericsson 1990; Harford 1980; Hovdenak 1987; Jaffe 1977; Lavö 1987; Lee 1968; Lustman 1987; Palmer 1980; Pelemans and Vantrappen 1976; Taneja 2004; Tijtgat 1975; Verhaegen 1974). Seven were conducted in the UK (Allison 1988; Cann 1984; Corbett 1980; Jaffe 1977; Lee 1968; Lustman 1987 and Palmer 1980); ten in the rest of Europe, three in the USA; one in India and one in South Africa.
All studies but six had fewer than 100 patients, with seven having 20 or fewer in the intervention arm (Allison 1988; Harford 1980; Hovdenak 1987; Lavö 1987; Taneja 2004; Tijtgat 1975; Verhaegen 1974). One study included 227 patients in total but only 46 in the loperamide group and 45 in the placebo group (Ericsson 1990). The remaining studies had between 152 and 614 patients (Dom 1974). Some of the studies were of crossover design, so fewer patients are required to achieve adequate power.
Study Design
There were seven crossover studies (Allison 1988; Cann 1984; Corbett 1980; Harford 1980; Palmer 1980; Pelemans and Vantrappen 1976; Verhaegen 1974) in which participants were allocated to receive both the intervention and control treatments during the course of the study, in a random order. Four of these studies (Allison 1988; Cann 1984; Corbett 1980; Palmer 1980) had either no washout period or it was not reported, in which case this was assumed to be none. One acute study had a washout period of 12 to 24 hours (Harford 1980). One longer term study had a washout period of 2 to 7 days, and the drugs were discontinued until severe diarrhoea returned (Verhaegen 1974). The other longer term study (Pelemans and Vantrappen 1976) had a washout period of 3 to 20 days (median 7 days). As the GDG had specified a washout period of one week minimum for maintenance studies, the latter three studies were included on the basis of washout period, but those with no washout were excluded from the analysis and transferred to the excluded studies table. The remaining studies had a parallel design.
The GDG had specified a minimum treatment period of four weeks for each intervention in the maintenance studies. One study had a treatment duration of one week (Barbezat 1979); one had a duration of at least 10 days or until relapse (Tijtgat 1975); one had at least 12 days (Verhaegen 1974), one had from 14 to 49 days, median 25 days (Pelemans and Vantrappen 1976); one had three weeks (Hovdenak 1987). The GDG decided to exclude Barbezat (1979); Tijtgat (1975), and; Verhaegen (1974) on this basis, and to accept Hovdenak (1987), and; Pelemans and Vantrappen (1976). The remaining studies had durations of seven weeks (Efskind 1995), 2 months (Taneja 2004) and 13 weeks (Lavö 1987).
Thus, 15 studies were included in the analysis: thirteen parallel (Amery 1975; Cornett 1977; Dettmer 1994; Dom 1974; Dreverman 1995; Efskind 1995; Ericsson 1990; Hovdenak 1987; Jaffe 1977; Lavö 1987; Lee 1968; Lustman 1987; Taneja 2004), and two crossover (Harford 1980; Pelemans and Vantrappen 1976).
Ten studies investigated the use of anti-motility agents for acute diarrhoea (Amery 1975; Cornett 1977; Dettmer 1994; Dom 1974; Dreverman 1995; Ericsson 1990; Harford 1980; Jaffe 1977; Lee 1968; Lustman 1987). Only one of these had some patients with IBS (Harford 1980). Five studies investigated the effectiveness of anti-motility agents for the treatment chronic diarrhoea in IBS.
Three studies had more than two arms. Amery (1975) compared diphenoxylate with loperamide and placebo. Dettmer (1994) and Dreverman (1995) compared two dose of loperamide with placebo, giving a total of 21 comparisons.
Setting: seven studies were in primary care (Amery 1975; Dom 1974; Dreverman 1995; Efskind 1995; Jaffe 1977; Lee 1968; Lustman 1987); three were in secondary care (Dettmer 1994; Lavö 1987; Taneja 2004) and the others did not report the setting.
Funding: four studies (Amery 1975; Cornett 1977; Dettmer 1994; Dreverman 1995) were from Janssen Pharmaceutica (manufacturers of Imodium, i.e., loperamide) and Efskind (1995) stated that Janssen Pharmaceutica provided the drug, monitored the study and gave statistical support. Lustman (1987) did not specify funding, but the corresponding author was employed by Gold Cross Pharmaceuticals, a division of GD Searle & Co Ltd (manufacturers of Lomotil). Three studies were funded by non-industry sources (Ericsson 1990; Harford 1980; Taneja 2004) and the others did not state their funding.
Population
Four studies were in patients with IBS (Efskind 1995; Hovdenak 1987; Lavö 1987; Taneja 2004), and two studies had some patients with IBS. The acute study, Harford (1980), had 4/15 patients with IBS; and the maintenance study, Pelemans and Vantrappen (1976), had 4/23; 18 of the remaining patients in the latter study had inflammatory bowel disease. In both studies, individual patient data were reported for the IBS subgroup, but these were not stratified before randomisation and the small numbers give uncertainty and likely potential for bias. The definition of IBS varied between studies: one included patients meeting the Rome II criteria (Taneja 2004), one met criteria defined by the authors that were similar to the Rome criteria (Efskind 1995), and in the other studies, the authors stated that the patients had IBS, with no further explanation.
Most studies included patients with diarrhoea predominance, but one study (Hovednak 1987) had a mixture of types: IBS-D (16); IBS-A with pain (21); IBS-A without pain (12); IBS-C (9). None of the studies stated that any participants had IBS as result of gastrointestinal infection. The majority of studies did not state the number of participants with bloating. One study had some patients with bloating (Efskind 1995).
Most of the studies did not describe symptom severity. Two studies stated that participants had symptoms of mixed severity (Efskind 1995; Hovednak 1987), one of which excluded patients with mild symptoms (Hovednak 1987).
The remaining studies were in patients who did not have IBS and these were treated as indirect evidence. Further details are given in the included studies table.
The age range of participants across the IBS studies was 18 to 70 years, with the mean age, where given, ranging from 31 to 43 years. No study particularly identified elderly participants. The indirect studies included patients aged 9 to 95 years, with four of the studies definitely including children: Amery (1975) included patients aged 9 to 82 years, with a median age of 31 years; Cornett (1977) had an age range of 11 to 84 years, with 8% patients in the age group 10 to 19 years; Dom (1974) had a range of 14 to 95 years with a median of 35 years; and Drevermann (1995) had patients aged 16 to 75 years.
Five studies had more women than men (Efskind 1995; Harford 1980; Lavö 1987; Lee 1968; Lustman 1987); two studies had about the same number of men and women (Dettmer 1994; Pelemans and Vantrappen 1976); one study examined only men (Taneja 2004) and the other indirect studies had more men than women. One study did not report the numbers of men and women (Ericsson 1990).
Interventions
The studies varied in the type of antimotility agent used:
- No studies examined codeine phosphate
- Seven acute studies gave the patients co-phenotrope (Amery 1975; Cornett 1977; Dom 1974; Harford 1980; Jaffe 1977; Lee 1968; Lustman 1987). The Amery study stated that they gave the patients 2.5mg diphenoxylate and that the contents of the capsules were identical to Lomotil (co-phenotrope)
- Twelve studies (seven acute) gave the patients loperamide (Amery 1975; Cornett 1977; Dettmer 1994; Dom 1974; Dreverman 1995; Efskind 1995; Ericsson 1990; Hovdenak 1987; Jaffe 1977; Lavö 1987; Pelemans and Vantrappen 1976; Taneja 2004)
- One acute study examined morphine (Lee 1968 used a kaolin and morphine mixture).
Ten of the included studies used antimotility agents for acute diarrhoea (Amery 1975; Cornett 1977; Dettmer 1994; Dom 1974; Dreverman 1995; Ericsson 1990; Harford 1980; Jaffe 1977; Lee 1968; Lustman 1987).
In the maintenance studies, no study allowed rescue medication, but patients were allowed to vary the dose of study drug in three studies (Efskind 1995; Lavö 1987; Pelemans and Vantrappen 1976). A fixed dose was used in the remaining studies.
Comparisons
The included studies covered the following comparisons:
- Ten comparisons of antimotility agent versus placebo:
- Four gave loperamide for acute episodes (Amery 1975; Dettmer 1994, in a dose of 1mg or 2mg; Dreverman 1995 in a dose of 1mg or 0.5mg; Ericsson 1990 2mg)
- Three gave loperamide for maintenance treatment (Efskind 1995; Hovdenak 1987; Lavö 1987).
- Three comparisons of different types of antimotility agent:
- Four studies compared loperamide and diphenoxylate for acute episodes (Amery 1975; Cornett 1977; Dom 1974; Jaffe 1977)
- One study compared loperamide and co-phenotrope for maintenance treatment (Pelemans and Vantrappen 1976)
- One study compared co-phenotrope with kaolin-and-morphine for acute episodes (Lee 1968).
- Two acute studies compared two doses of loperamide (Dettmer 1994: 1mg versus 2mg; Dreverman 1995 1mg versus 0.5mg)
- One maintenance study compared loperamide with yoga (Taneja 2004).
METHODOLOGICAL QUALITY
The results of the quality assessment for included trials are shown in Appendix D. The method of randomisation was reported in none of the studies. Allocation concealment was reported in one study (Pelemans and Vantrappen 1976), which reported a partially adequate method in which bottles of the drug were marked with the patient’s code number and contained identical capsules.
All the studies reported that the patients were blinded to the interventions except for three. Jaffe (1977) reported that the drugs were presented in their normal marketed form and as they are dissimilar in appearance and had different dose regimens, no attempt was made to blind participants or investigators. Lee (1968) did not report blinding and the treatments used were dissimilar: Lomotil-with-neomycin was used at the recommended dose of 4 tablets at the start of therapy then 2 tablets every 6 hours, while the kaolin-and-morphine mixture was used at 2 tablespoons at the start of treatment then 1 tablespoon every 6 hours. Taneja (2004) compared loperamide with yoga and this was not blinded. Only two studies reported a sample size calculation (Dettmer 1994; Ericsson 1990).
Most studies included in the review demonstrated baseline comparability of the groups, but two studies were not comparable at baseline for the age of the patients (Amery 1975 had significantly older patients in the loperamide group; Lavö 1987 had significantly younger patients in the loperamide group). The GDG did not regard these differences to be important.
Five acute diarrhoea studies reported no withdrawals (Amery 1975; Cornett 1977; Dom 1974; Jaffe 1977; Lee 1968). One of the 13 patients allocated to loperamide in the Taneja (2004) study could not attend for the final assessment but the nine treated with yoga all attended. Dettmer (1994) reported missing data for 7% (13/230) of patients. Dreverman (1995) reported missing data for 3% (8/242) of patients. Lustman (1987) reported missing data for 4% (7 of 152) of patients.
In the Ericsson (1990) acute diarrhoea study, 46 patients were allocated to the loperamide group and 45 to the placebo group. Of these, 6 and 4 patients respectively dropped out and were excluded from the efficacy analysis. In addition, those who were non-compliant with medication (13 and 14 patients respectively) were also excluded from the analysis. This led to the final numbers being 27 for each group (i.e. missing data for 41% in each group). This raises the potential for bias in this study.
Two maintenance studies reported that more than 20% of patients in at least one arm (or overall) were not analysed or were lost to follow-up (attrition bias). Efskind (1995) reported 23% (21/90) missing overall, but 8 of these did not arrive at the start of the trial (thus drop-outs related to treatment were 12/90, i.e. 13%). Pelemans and Vantrappen (1976) had 26% (6/23) missing overall for the stool frequency outcome measure, but none of the IBS patients were missing.
Two of the studies (Harford 1980; Pelemans and Vantrappen 1976) each gave individual patient data for 4 IBS patients. This was a within-trial subgroup analysis and stratification had not taken place. The GDG decided to consider the results for all patients in the Harford (1980) acute study, but decided that the Pelemans and Vantrappen (1976) maintenance study had too few IBS patients and would be misleading, so this study was not considered further.
The risk of bias was assessed for each included study and two studies were excluded from the analysis: Pelemans and Vantrappen (1976) was excluded on the basis of wrong population, and; Ericsson (1990) was considered to have too high a drop-out rate. None of the other studies were considered to be at risk of bias, although the inclusion of children in some studies was noted.
RESULTS
I. TREATMENT FOR ACUTE EPISODES OF DIARRHOEA
For the treatment of acute diarrhoea the GDG simply wished to know whether anti-motility agents were effective in stopping diarrhoea, regardless of its cause. Therefore only outcomes relating to bowel habit are reported.
A. Anti-motility agent versus placebo
Three studies gave co-phenotrope for acute episodes (Amery 1975; Harford 1980; Lustman 1987).
- Amery (1975) gave 5 mg diphenoxylate per day (1 tablet twice a day) for 24 hours
- Harford (1980) gave 10 or 20 mg per day (1 or 2 tablets 4 times a day) for 3 days.
- Lustmann (1987) gave 10 mg initially (4 tablets) then 20mg per day (2 tablets four times per day) for 3 days.
The recommended initial dose for co-phenotrope is initially 10 mg (4 tablets) then 20mg per day (2 tablets four times a day), thus the Amery (1975) study can be considered to have a low dose of diphenoxylate.
Three studies gave loperamide for acute episodes compared with placebo (Amery 1975; Dettmer 1994, 2 doses; Dreverman 1995, 2 doses).
- Amery (1975) gave 4 mg loperamide per day (1 tablet twice a day) for 24 hours
- Dreverman (1995a) gave 1mg loperamide (2 tablets) initially then up to 3.5mg (7 tablets) per day for 3 days
- Dreverman (1995b) gave 2mg loperamide (2 tablets) initially then up to 7mg (7 tablets) per day for 3 days
- Dettmer (1994a) gave loperamide in a slow release formulation, 2 mg initially (2 tablets) then up to 8 mg (8 tablets) per day for 3 days
- Dettmer (1994b) gave loperamide in a slow release formulation, 4 mg initially (2 tablets) then up to 16 mg (8 tablets) per day for 3 days.
The recommended dose is 4 mg initially (2 tablets) followed by 2 mg (1 tablet) after each loose stool for up to 5 days.
We noted that all of the loperamide studies were funded by Janssen Pharmaceutica, manufacturers of Imodium (i.e. loperamide).
A1. Co-phenotrope versus placebo
i. Number of patients with improvement in bowel habit
One study (Amery 1975) reported the number of patients without recurrence (unformed stools) after 1, 2, 4 and 24 hours. There was no significant difference between diphenoxylate and placebo at any duration.

Figure 1Acute diarrhoea – co-phenotrope
ii. Stool frequency
Harford (1980) recorded stool frequency averaged over 3 days of co-phenotrope or placebo in a crossover study in 15 patients; individual patient data were given. Results are reported for all patients and for the IBS subgroup separately. For all patients, there was a statistically significant difference in stool frequency of −2.29 stools per day (95%CI −4.47, −0.11), favouring co-phenotrope. For the IBS subgroup, the effect was not statistically significant, but the confidence interval was wide.

Figure 2Stool frequency
Lustman (1987) reported the change in the median number of bowel actions (24 hours prior to start of therapy minus first 24 hours of treatment). This fell from 5 in each group to 3 on diphenoxylate, compared to 4 on placebo, which was a statistically significant difference (p=0.046).
iii. Time to recurrence of unformed stools
Amery (1975) also reported the median time to first recurrence of unformed stools. This was 2 hours for both diphenoxylate and placebo groups, i.e. no significant difference (p=0.48).
Lustman (1987) reported the median time to the last loose or watery stool, which was 25 hours on diphenoxylate compared with 30 hours for placebo (not statistically significant).
Lustman (1987) also reported the median time (hours) to the start of an interval of at least 12 hours between bowel actions: 14 hours for diphenoxylate vs. 24 hours for placebo, p=0.025.
iv. Stool weight
Harford (1980) recorded stool weight averaged over 3 days of co-phenotrope or placebo in a crossover study in 15 patients. The results are reported for all patients and for the IBS subgroup separately. The confidence interval was too wide to determine if there was a difference for all patients and there was no statistically significant difference for the IBS patients.

Figure 3Stool weight
A2. Loperamide versus placebo
i. Number of patients with improvement in bowel habit
Amery (1975) reported the number of patients without recurrence (unformed stools) after 1, 2, 4 and 24 hours. Dettmer (1994) also recorded the number of patients without diarrhoea at 3 days for the two loperamide doses combined.
At 1 and 2 hours Amery (1975) showed no significant difference between loperamide and placebo. After 4 hours there were significantly fewer patients with unformed stools in the loperamide group compared to placebo, with a number needed to treat of 5 (95%CI 3, 17), for a control group risk of 36%. After 24 hours, loperamide showed fewer patients with unformed stools, with borderline significance, but the confidence interval was fairly wide. At 72 hours, Dettmar (1994) showed a small statistically significant difference between loperamide and placebo, with an NNT of 7 (95%CI 4, 34), for a control group risk of 73%.

Figure 4Acute diarrhoea – loperamide
Dreverman (1995) reported the number of patients achieving first relief (the start of a 24 hour period during which no more than 1 pasty stool and no watery or loose stools were passed). There were statistically significantly more patients achieving relief for each dose of loperamide compared with placebo, but the confidence intervals are wide.

Figure 5
ii. Time taken to first relief of symptoms
Two studies reported the median time to first relief of symptoms, but different doses were used.
- 0.5 mg versus placeboDreverman (1995) in 156 patients: 20 hours 15 minutes for loperamide 0.5mg versus 24 hours 50 minutes for placebo (p=0.012), i.e. statistically significantly in favour of loperamide
- 1 mg versus placebo (2 studies)
- Dettmer (1994): 22.4 hours for loperamide 1mg versus 30 hours for placebo
- Dreverman (1995) in 158 patients: 15 hours 30 minutes for loperamide 1mg versus 24 hours 50 minutes for placebo (p=0.003), i.e. highly statistically significant, in favour of loperamide
- 2 mg versus placebo
- Dettmer (1994): 22.1 hours for loperamide 1mg versus 30 hours for placebo.
In Dreverman (1995), the median time to complete relief of symptoms was 26 hours 30 minutes for loperamide 1mg; 25 hours 40 minutes for loperamide 0.5mg; and 34 hours 15 minutes for placebo (p=0.041 for 0.5 mg and 0.044 for 1mg).
iii. Time to first recurrence
Amery (1975) also reported the median time to first recurrence of unformed stools. For the loperamide and placebo groups, this was 24, and 2 hours respectively. The p value for the difference between loperamide and placebo was p=0.016, i.e. statistically significant.
B. Anti-motility agent dose 1 versus dose 2
i. Number of patients with improvement in bowel habit
Dreverman (1995) reported the number of patients achieving first relief. There was no significant difference between the doses and the confidence interval was fairly wide.

Figure 6
ii. Median time to first relief of symptoms
- 0.5 mg versus 1 mgDreverman (1995): 20 hours 15 minutes for loperamide 0.5mg versus 15 hours 30 minutes for loperamide 1mg
- Dettmer (1994): 22.4 hours for loperamide 1mg versus 22.1 hours for loperamide
In Dettmar (1994), the median time to complete relief of symptoms was 27.55 hours for loperamide 1mg and 25.00 hours for loperamide 2mg, with no significant difference between dose groups.
C. Anti-motility agent type 1 versus type 2
C1. Co-phenotrope versus loperamide
Four studies compared co-phenotrope and loperamide for acute episodes (indirect population) (Amery 1975; Cornett 1977; Dom 1974; Jaffe 1977). Although different doses were compared across studies, the proportions of drugs were the same.
- Amery (1975): 5 mg diphenoxylate per day (1 tablet twice a day) versus loperamide 4 mg (2 mg twice a day) for 24 hours.
- Cornett (1977): initially 5mg (2 capsules) co-phenotrope then up to 20 mg per day versus 4 mg (2 capsules) loperamide initially, then up to 16 mg per day
- Dom (1974): initially 5 mg (2 capsules) co-phenotrope then up to 25 mg per day versus 4 mg loperamide intitially (2 capsules) then up to 20 mg per day
- Jaffe (1977): initially 10 mg (4 capsules) co-phenotrope then 20 mg per day (2 capsules × 4) versus 4 mg (2 capsules) loperamide initially, then up to 16 mg per day.
We noted that Amery (1975) and Cornett (1977) were funded by Janssen Pharmaceutica, manufacturers of Imodium (i.e. loperamide).
i. Number of patients with improvement in bowel habit
Three studies reported the number of patients with no unformed stools at different durations: Amery (1975) reported the number of patients without recurrence (no unformed stools) after 1, 2, 4 and 24 hours; Dom (1974) recorded the same outcome after 24 hours and 2 and 3 days; Cornett (1977) recorded the number of patients with unformed stools, from which we calculated the number with no unformed stools after 24 hours and 2 and 3 days. Jaffe (1977) reported the number of patients not reaching a ‘cure’ after 4 days (figure 10). At 1 and 2 hours there were statistically significantly more patients who were diarrhoea free for the loperamide group compared to the diphenoxylate. NNTs are 4 (95%CI 3, 12) and 5 (9%CI 3, 34). There was no significant difference at 4 hours. At 24 hours, meta-analysis of three studies showed statistically significantly more patients had no unformed stools for the loperamide group; RR 0.78 (95%CI 0.62, 0.98); with some heterogeneity (I2=47%, p=0.15). The NNT was 20 (95%CI 10, 100) for a control group rate of 21 to 41%. At 2 and 3 days there was still a statistically significant effect favouring loperamide, but there was significant heterogeneity for the 3 day meta-analysis. We note, however, that some of these trials were industry sponsored.


Figure 7Number of patients with no unformed stools
ii. Time taken to first stools
Jaffe (1977) reported the mean time to the 1st, 2nd, and 3rd motion. In all cases the confidence intervals were wide.


Figure 8
Amery (1975) reported the median time to first recurrence of unformed stools. For the loperamide and diphenoxylate groups, this was 24 and 2 hours respectively. This was a statistically significant difference (p=0.024), favouring loperamide. We note, however, that this was an industry funded study.
iii. Stool frequency
Jaffe (1977) reported the stool frequency in the first 6 hours; first 12 hours; first 24 hours; and first 48 hours following the first dose of the medication. In all cases the confidence intervals were wide, so conclusions were not drawn.


Figure 9
Cornett (1977) also reported the frequency of unformed stools over 72 hours. From a mean of 9.14 unformed stools before treatment, patients on diphenoxylate reduced to 5.47 (a fall of 3.67) while those on loperamide fell from 8.75 to 4.38 (a fall of 4.37). No standard deviations were given.
Dom (1974) reported the change in the mean number of unformed stools in a 72 hour period (from 8.06 before treatment with diphenoxylate and 8.02 in the loperamide group to 3.68 and 2.65 respectively, i.e. a fall of 4.38 and 5.37 respectively, this was statistically significantly in favour of loperamide (p=0.011).
iv. Time to cure
Jaffe (1977) reported the mean time to cure. There was a wide confidence interval so conclusions were not drawn.
C2. Co-phenotrope versus morphine
i. Number of patients with normal stool frequency
One study (Lee 1968) compared cophenotrope-with-neomycin to kaolin-and-morphine. This study reported the number of patients with abnormal stool frequency after 12 hours, 24 hours, 2 days, 3 days and 4 days. We have reported the number of patients with normal stool frequency. There was a statistically significant difference favouring co-phenotrope at durations up to 48 hours. At 12 hours the NNT was 4 (95%CI 3, 6), for a control group risk of 14%.

Figure 11
ii. Number of patients with normal stool consistency
One study (Lee 1968) compared cophenotrope-with-neomycin to kaolin-and-morphine. This study reported the number of patients with abnormal stool consistency after 12 hours, 24 hours, 2 days, 3 days and 4 days. We have reported this as the number of patients with normal stool consistency. There was a statistically significant effect, favouring co-phenotrope at 12 hours (RR 3.49 (95%CI 1.60, 7.60); NNT 5 (95%CI 4, 10), for a control group risk of 9%. Otherwise there was no significant difference between interventions.

Figure 12
II. MAINTENANCE TREATMENT FOR CHRONIC DIARRHOEA
A. Antimotility agent versus placebo
Three studies gave loperamide for maintenance treatment (Efskind 1995; Hovdenak 1987; Lavö 1987). All were in patients with IBS, although Hovdenak (1987) stated that the patients had different types of IBS, including IBS-C. Subgroup analyses were presented, but these did not constitute stratification before randomisation. This study also had a duration of 3 weeks, but the GDG agreed that this was acceptable. We noted that Efskind (1995) was industry supported (by Janssen Pharmaceutica, the manufacturers of loperamide).
The dose in Hovdenak (1987) was fixed at 4 mg at night, whilst the patients were able to adjust the dose in the other two studies. Both started the patients on 2 mg at night, which was increased to 6 mg (Efskind 1995) or 8 mg (Lavö 1987) as required.
1. Global symptoms
a. Number of patients with improvement in global symptoms
One study (Hovdenak 1987) reported the number of patients with improvement in global symptoms after 3 weeks. The study reported results for three of the different IBS subgroups (IBS-C results were not reported). The exclusion of the IBS-C results breaks the randomisation, so these are post-hoc subgroups, but still gives some information. We have grouped together the results for IBS-D, IBS-A (with pain), IBS-A (without pain), and the separate subgroups. There was a statistically significant improvement in symptom score for the 3-type IBS group and for the IBD-D group alone, although the confidence interval for the latter was wide. The relative risks were 2.00 (95%CI 1.15, 3.48) and 4.00 (1.20, 13.28) respectively. These corresponded to a number needed to treat of 3 (95%CI 2, 8) and 2 (95%CI 1, 3), for control group rates of 39% and 25% respectively.

Figure 13
b. Global symptom improvement score
One study (Lavö 1987) reported a subjective overall response for the whole 13 week study period, which was said to statistically significantly favour loperamide (p<0.03).
2. Individual symptoms
a. Pain
i. Number of patients with less pain
Two studies reported the number of patients with less pain, one (Hovdenak 1987) after 3 weeks in a range of different IBS subgroups; and the other (Lavö 1987) after 13 weeks in patients with IBS-D only. For the former study we grouped together the results for IBS-D, IBS-A (with pain), IBS-A (without pain) and also reported these separately.
The confidence intervals were wide for each study. Meta-analysis of the 3-type IBS group with the Lavö study, in 70 patients gave a statistically significant difference between interventions, favouring loperamide; RR 2.60 (95%CI 1.02, 6.61) but there was some heterogeneity (I2=48%, p=0.17). The NNT was 5 (95%CI 3, 25), for a control group rate of 8 to 17%.
ii. Number of patients with increased pain
Two studies reported the number of patients with more pain, one (Hovdenak 1987) after 3 weeks in a range of different IBS subgroups; and the other (Lavö 1987) after 13 weeks in patients with IBS-D only. For the former study we grouped together the results for IBS-D, IBS-A (with pain), IBS-A (without pain) and also reported these separately.
The confidence intervals were wide for each study. Meta-analysis of the 3-type IBS group with the Lavö (1987) study, in 70 patients, gave a statistically significant difference between interventions, favouring loperamide; RR 0.38 (95%CI 0.15, 0.96) and there was no heterogeneity (I2=0%), although the confidence interval was wide. The NNT was 5 (95%CI 3, 25), for a control group risk of 22 to 67%. For the meta-analysis of 40 patients with IBS-D, the difference was still statistically significant with no heterogeneity; RR 0.36 (95%CI 0.14, 0.96), i.e. 3 times more risk of pain for the placebo group (Figure 14). The NNT was 3 (95%CI 2, 13) for a control group risk of 38 to 67%, but the confidence interval was wide.

Figure 14
Number of patients with less pain.
iii. Number of days with pain
One study (Hovdenak 1987) recorded the number of days with pain over 3 weeks. In the subgroup of IBS-A with pain, there was a statistically significant mean difference of 6.1 days between loperamide and placebo (p<0.01), favouring the former.
iv. Pain score
One study (Efskind 1996) in 69 patients reported no significant difference between interventions in the change from baseline in abdominal pain measured on a visual analogue scale. This was the case for both their 2 week dose adjustment period and for 5 weeks of fixed dose loperamide or placebo. However, the study found significantly more pain at night for the loperamide group (p<0.05 for both periods). In contrast, another study in 21 patients (Lavö 1987) found a statistically significant difference in pain score, favouring loperamide (p<0.05).
b. Bowel habit
i. Number of patients with improved bowel habit (frequency)
Two studies reported the number of patients with improved stool frequency, one (Hovdenak 1987) after 3 weeks in different IBS subgroups; and the other (Lavö 1987) after 13 weeks in patients with IBS-D only. For the former study we grouped together the results for IBS-D, IBS-A (with pain), IBS-A (without pain) and also reported these separately.
Meta-analysis of the 3-type IBS group with the Lavö study, in 70 patients gave a statistically significant difference between interventions, favouring loperamide; RR 2.38 (95%CI 1.53, 3.70) and there was no heterogeneity (I2=0%). This corresponded to an NNT of 2 (95%CI 2, 4) for a control group risk of 25–43%. For the meta-analysis of 40 patients with IBS-D, the difference was still statistically significant with no heterogeneity; RR 2.83 (95%CI 1.43, 5.63). This corresponded to a number needed to treat of 2 (95%CI 2, 4) for a control group risk of 25–38% (figure 15). Statistically significant improvements were also achieved for the IBS-A group, which had an NNT of 2 (95%CI 2, 4), for a control group risk of 45%. Confidence intervals for these meta-analyses were fairly wide.

Figure 15
Number of patients with more pain.

Figure 16Improvement in stool frequency
ii. Number of patients with improved bowel habit (consistency)
The same two studies reported the number of patients with improved stool consistency (Hovdenak 1987; Lavö 1987). Meta-analysis of the 3-type IBS group with the Lavö (1987) study, in 70 patients gave a statistically significant difference between interventions, favouring loperamide; but there was significant heterogeneity (I2=69%; p=0.07). This heterogeneity may possibly be explained by differences in the study duration or the intervention dose, both of which were higher for the Lavö (1987) study. For the meta-analysis of 40 patients with IBS-D, the difference was still statistically significant with some heterogeneity (I2=59%; p=0.12). Random effects analysis showed wide confidence intervals and neither analysis was statistically significant. Statistically significant improvements were also achieved for the combined IBS-A group; RR: 2.10 (95%CI 1.23, 3.58), which corresponded to an NNT of 3 (95%CI 2, 5) for a control group risk of 44%.

Figure 17Improvement in stool consistency
iii. Stool scores
Two studies (Efskind 1996 (n=69) and Lavö 1987 (n=21)) reported a stool score measure for both consistency and frequency. Each found a statistically significant improvement in stool consistency (p<0.002 and p<0.001) respectively, favouring loperamide. Efskind (1996) also found a statistically significant difference over 7 weeks for frequency (p<0.05) but Lavö (1987) found no significant difference. Lavö (1987) found a highly significant difference in the number of formed stools, favouring the loperamide group.

Figure 18Number of formed stools
iv. Number of patients with improvement in urgency
One study (Lavö 1987) in 21 patients reported the number of patients with less urgency and found this to be significantly greater for the loperamide group, although the confidence interval was wide; RR 3.00 (95%CI 1.07, 8.43). This corresponds to an NNT of 2 (95%CI 2, 7), for a control group rate of 25%.

Figure 19Number of patients with less urgency
3. Adverse effects
Only one small study in 24 patients reported the number of patients with adverse effects (Lavö 1987). In all cases there are very wide confidence intervals and conclusions cannot be drawn.

Figure 20Adverse effects
B. Anti-motility agent type 1 versus Anti-motility agent type 2
One study compared different types of anti-motility agent: Pelemans and Vantrappen (1976) compared co-phenotrope and loperamide for 14 to 49 days in a crossover trial in 23 patients. Only 4 of the patients had IBS, 18 of the rest had inflammatory bowel disease and the GDG considered this population to be too indirect to be useful, even though this was the only maintenance study considering co-phenotrope.
C. Antimotility agent versus yoga
1. Bowel symptom score
Taneja (2004) reported bowel symptom scores 1 and 2 months after starting either loperamide or yoga. Patients were asked the following questions: Is the frequency of stools >3 times per day? Is the frequency of stools <3 times per week? Are the stools lumpy or hard? Is there straining? Are the stools loose or watery? Is there urgency to pass the stools? Are the stools mucoid? For each symptom, if present >25% of the time, a positive answer scored 1 and a negative answer 0, so the range is 0–7. Both groups reduced their scores from baseline (loperamide 4.08 (SD 0.9); yoga 3.77 (SD 1.2)), but there was no significant difference between the groups.

Figure 21Bowel symptom score
Adverse Effects
Two RCTs with adverse effects data were identified (Lavo 1987; Cann 1984). One was a crossover trial which should be treated with caution (Cann 1984). No clear trend could be identified from these two studies but abdominal cramps, dizziness, drowsiness, and skin reactions including urticaria; paralytic ileus and abdominal bloating have been reported (BNF 2007).
ECONOMIC LITERATURE FOR ANTIMOTILITY AGENTS
No relevant health economic analyses were identified on the cost-effectiveness of antimotility agents in the treatment of IBS.
COST-EFFECTIVENESS ANALYSIS FOR ANTIMOTILITY AGENTS
This section describes the health economic analysis undertaken to inform recommendations on the use of antimotility agents as a long-term maintenance therapy in IBS. The general methods used in the economic analysis for all management interventions are described in detail in Chapter 5 and the model inputs and assumptions relevant to this particular intervention are described below.
- Loperamide was the only intervention considered by the economic model as it was the only intervention which had evidence of clinical effectiveness for longer-term maintenance use.
- Separate efficacy estimates were available for subgroups of patients with IBS-D and patients with IBS-A with pain or IBS-A without pain. Loperamide was statistically significantly better than placebo in achieving an improvement in global symptoms for all three subgroups combined but was also statistically significantly better for the IBS-D subgroup but not for the IBS-A subgroups when considered separately. However, the confidence intervals for the subgroups were wide due to the small number of patients in each subgroup. The combined effectiveness estimate for IBS-D and IBS-A (with and without pain) was used in the basecase.
Modelled response rates
In the basecase scenario the response rate of 45% in the no treatment arm is taken from the Mearin (2004) cohort study. This represents the group of patients for whom we would expect an improvement in symptoms without any specific intervention. The RR of response for antimotility vs placebo is 2.00; therefore the response rate in the intervention arm is 90% (=45% × 2.00) giving an absolute difference in response between the intervention and no treatment arms of 45% (=90%-45%) during the 1st month.
We have also considered an alternative scenario in which no patient in the comparator arm achieves an improvement in symptoms but there is still a 45% chance of response for the intervention arm.
Table 1Intervention specific parameters – anti-motility agents
| Description | Value | Evidence | |
|---|---|---|---|
| RR of response for intervention vs placebo | 2.00 | Meta-analysis of RCT evidence for improvement in global symptoms | |
| Maximum number of switches considered | 0 | Limited by number of effective interventions | |
| Drug costs | |||
| Intervention | Dose per day | Cost per month* (assuming lowest cost preparation) | |
| Loperamide | 4mg | £2.29 | |
- *
British National Formulary (Joint Formulary Committee 2007)
In the basecase analysis, treatment with loperamide for 100 patients with IBS-D or IBS-A results in an additional 1.60 QALYs but costs an additional £3,055 over a 6 month time frame, compared to no treatment, giving a cost per QALY of £1,914. The basecase analysis assumes that the intervention is used on a daily basis, meaning this estimate may be more relevant to patients with IBS-D. The cost-effectiveness of using loperamide on 25% to 75% of days is considered in the sensitivity analysis, and this estimate may be more relevant to patients with IBS-A whose primary symptom may vary from constipation to diarrhoea.
These results are an estimate of the cost-effectiveness over the first 6 months after initiating treatment with loperamide. The cost per QALY for continuing treatment with loperamide beyond 6 months is lower than the cost per QALY during the initial 6 months provided that treatment is reviewed every 6 months and discontinued in patients who no longer experience a therapeutic benefit, either due to lack of effectiveness or a change in their symptom profile.
The probabilistic sensitivity analysis considered the uncertainty in the basecase estimate of cost-effectiveness due to uncertainty in the parameters used to estimate the cost-effectiveness. The CEAC presented in Figure 22 shows the probability that the cost per QALY for loperamide compared to no treatment is under various cost per QALY thresholds. For example, it shows that there is an 83% probability that loperamide has a cost per QALY of under £5,000 compared to no treatment and a 98% probability that it has a cost per QALY under £20,000. However, it should be noted that these estimates only consider the uncertainty in cost-effectiveness due to the accuracy of several input parameters and they do not reflect general uncertainty around the assumptions made in the model. The uncertainty from these assumptions was explored in the univariate sensitivity analysis.
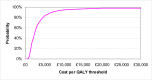
Figure 22
Cost-effectiveness acceptability curve for loperamide compared to no treatment.
Univariate sensitivity analysis
Maintaining the 45% difference in response between the two arms but reducing the response rate in the no treatment arm from 45% to zero marginally decreased the cost per QALY to £1,593.
We carried out a threshold analysis to determine whether antimotility therapy is still cost-effective for lower gains in health related quality of life. In the basecase it is assumed that patients who respond to therapy accumulate 0.071 QALYs more per annum than patients who do not respond. For comparison, a gain of 0.135 QALYs would represent a complete remission of IBS symptoms. If the QALY gain associated with a response to therapy was reduced to 0.006 QALYs, then the cost per QALY of providing antimotility therapy would be above £20,000 per QALY.
The basecase analysis assumes that the lowest cost preparation is prescribed. If, the highest cost preparation (Imodium syrup) is prescribed at the same dose, then the cost per QALY is increased to £3,173. If the dose is increased to the maximum dose of 16mg per day, which is double the highest dose used in the maintenance therapy trials, and the highest cost preparation is prescribed, then the monthly cost of loperamide rises to £23.85, but the cost per QALY is still well below £20,000 per QALY at £9,311.
If a patient takes a therapy on an as needed basis, it is assumed that they take the therapy on days when their quality of life is significantly affected by their IBS symptoms but not on days when their symptoms are less severe. They only accrue QALY benefits and drug costs on the days they take the therapy. However, it is still necessary to assess all patients for response after 1 month of initiating therapy. This means that it is less cost-effective to initiate therapy in patients who use the therapy on fewer days as the monitoring costs are just as high but the benefits are lower. This is shown by the cost per QALY of £5,297 for patients who take the therapy on 25% on days.
Table 2Sensitivity results for loperamide compared to no treatment for 100 patients with IBS-D or IBS-A
| Scenario | No Treatment | Intervention | Incremental | ||
|---|---|---|---|---|---|
| Cost | QALY | Cost | QALY | Cost per QALY | |
| Basecase | £0 | 1.60 | £3,055 | 3.19 | £1,914 |
| No response in no treatment arm | £0 | 0 | £2,542 | 1.60 | £1,593 |
| Response rate in no treatment arm from RCTs | £0 | 1.69 | £3,117 | 3.38 | £1,842 |
| Treatment used 75% of days | £0 | 1.60 | £2,741 | 2.79 | £2,290 |
| Treatment used 50% of days | £0 | 1.60 | £2,428 | 2.39 | £3,042 |
| Treatment used on 25% of days | £0 | 1.60 | £2,114 | 2.00 | £5,297 |
| Half of treatment obtained over the counter | £0 | 1.60 | £2,428 | 3.19 | £1,521 |
| Higher cost formulations (same dose) | £0 | 1.60 | £5,066 | 3.19 | £3,173 |
| High utility gain of 0.135 | £0 | 3.02 | £3,055 | 6.04 | £1,011 |
| Threshold analysis on lowest utility | A cost per QALY of £20,000 is reached when the QALY gain associated with responding to treatment lies between 0.006 and 0.007 | ||||
EVIDENCE STATEMENTS
For this review, the evidence was assessed using the GRADE process and tables are shown in Appendix F. The following evidence statements are derived from the GRADE tables.
- In studies for short-term symptom relief of diarrhoea (including people with IBS) for loperamide versus placebo there is fair evidence showing a significant number of patients without unformed stools.
- In studies for short-term symptom relief of diarrhoea (including people with IBS) for co-phenotrope (lomotil) compared with placebo there is:
- Limited evidence showing significant improvement in stool frequency
- Fair evidence showing no significant difference in the number of patients without unformed stools
- In studies for short-term symptom relief of diarrhoea (including people with IBS) for loperamide versus co-phenotrope (lomotil) there is:
- Fair evidence showing a significant improvement in the number of patients without unformed stools.
- Good evidence to show a significant improvement in stool score.
- In studies for short-term symptom relief of diarrhoea (including participants with IBS) for co-phenotrope (lomotil) compared with Kaolin and Morphine there is fair evidence showing a significant improvement in stool consistency for co-phenotrope.
- In studies for longer-term maintenance treatment of diarrhoea (including participants with IBS) for loperamide versus placebo there is:
- Limited amount of fair quality evidence showing a highly significant improvement in global symptoms
- Limited evidence showing significant improvement in pain and bowel habit.
- In studies for longer-term maintenance treatment of diarrhoea (including people with IBS) for loperamide versus yoga there is a limited amount of weak evidence showing no significant difference in bowel score.
- There is no clear evidence on adverse effects for antimotility agents.
HEALTH ECONOMIC STATEMENT
Evidence from a decision analytic model showed that loperamide is cost-effective as a long-term maintenance therapy for individuals with diarrhoea provided that response is assessed after the first month and every 6 months thereafter and treatment is discontinued in individuals for which it provides no therapeutic benefit.
GDG DISCUSSION
The GDG noted that, despite the small number of trials, loperamide is widely used and accepted as clinically effective for the treatment of diarrhoea in people with IBS. The GDG noted that it was good practice to titrate the dose of loperamide, according to symptom response, with the aim of achieving a well formed stool (Bristol Stool Form Scale type 4). In certain situations the daily dose of loperamide required may exceed 16 mg, and the GDG noted that this is an out of licence dose.
EVIDENCE TO RECOMMENDATIONS
The GDG took into consideration the clinical and cost effective evidence and their experience of the widespread use of loperamide. They noted the lack of evidence about adverse effects, but did not consider this to be a significant factor in practice. The GDG wished to encourage primary care clinicians to advise people with IBS of the need to titrate doses and the method of doing so.
RECOMMENDATION
Loperamide should be the first choice of antimotility agent for diarrhoea in people with IBS2.
RECOMMENDATION
People with IBS should be advised how to adjust their doses of laxative or antimotility agent according to the clinical response. The dose should be titrated according to the stool consistency, with the aim of achieving a soft, well-formed stool (corresponding to Bristol Stool Form Scale type 4).
8.3. Antispasmodics
SELECTION CRITERIA
The selection criteria described in the general methodology section were used, but some were specific to the antispasmodics review and are reported below.
Types of studies
The GDG decided that the washout period for this review should be at least one week. Trials with shorter washout periods were not included in the analysis.
Types of intervention
Studies were to include the following interventions:
- Anti-muscarinic agents:
- Atropine (synonyms: hyoscyamine)
- Dicycloverine (synonyms: dicyclomine, diethylaminocarbethoxybicyclohexyl hydrochloride; trade name: Merbentyl®)
- Hyoscine (synonyms: scopolamine; trade name: Buscopan®)
- Propantheline (Trade name: Pro-Banthine®).
- Direct-action smooth muscle relaxants:
- Alverine (Trade name: Spasmonal®)
- Mebeverine: includes modified release and conventional drug delivery (Trade name: Colofac®)
- Peppermint oil: includes modified release and enteric coated (Trade names: Colpermin®; Mintec®).
Studies with interventions not available in the UK were included in the review. These studies were listed, but not included in the sections on analysis or characteristics of studies.
The following comparisons were included
- Antispasmodic versus placebo (or nothing)
- Antispasmodic type 1 versus type 2
- Antispasmodic dose 1 versus dose 2
- Antispasmodic + another intervention versus the other intervention alone
- Antispasmodic modified release versus conventional delivery
- Duration of treatment 1 versus duration 2.
NB: In spite of the large placebo effect associated with IBS, comparisons with no treatment were included.
The antispasmodics review was concerned with both longer term maintenance treatment and short term symptom relief.
The GDG decided that there should be a minimum duration of treatment of four weeks for maintenance in this review. Studies of shorter durations were included but dealt with in sensitivity analyses.
Subgroup analyses
We planned to carry out subgroup analyses by type of antispasmodic (antimuscarinic and direct-acting smooth muscle relaxants); dose; mode of delivery (modified release/conventional), and; duration of intervention.
Search strategy for identification of studies
The initial search identified a Cochrane Review (Quartero 2005, Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome). Searches were partly based on the terms in this review. Searches were performed on the following core databases: MEDLINE, EMBASE, CINAHL and The Cochrane Library (1966 to current day with guidance from the GDG). Additional databases were not searched for this review. The search strategies are listed in Appendix B.
The titles and abstracts identified by the search strategy were assessed. One-hundred and three were identified as being potentially relevant to the review and these papers were retrieved in full. Twenty-three studies met the inclusion criteria for the review. The reference lists for each of the retrieved studies were inspected for further potential papers, but none were identified. The 80 excluded studies are listed in Appendix E, along with the reasons for exclusion
DESCRIPTION OF STUDIES INCLUDED IN THE REVIEW
The Cochrane review recently published on this topic did not cover all the comparisons required for the guideline, as only comparisons with placebo were included. The interventions in the Cochrane Review also extended to drugs not licensed for use in the UK, and the subdivision by types of antispasmodic was different from that determined by the GDG. In addition, crossover trials were excluded from the Cochrane review unless first-period only results were given. We also did not agree with some of the other inclusions/exclusions. A simple update of the Cochrane review was therefore not appropriate to the needs of the guideline and the review was instead used mainly as a reference. Studies excluded from the Cochrane review as non-placebo comparisons or crossover trials were assessed for inclusion in the guideline review.
This review relates only to those studies using interventions licensed for use in the UK. The following 38 studies were therefore eliminated from the review:
- Cimetropium bromide (Centonze 1988; Dobrilla 1990; Passaretti 1989; Piai 1985; Piai 1987)
- Fenoverine (Galeone 1992)
- Octatropine methylbromide (Barbara 1979)
- Octilonium bromide (Baldi 1992; Capurso 1984; Capurso 1992a; D’Arienzo 1980; Defrance 1991; Ferrari 1986)
- Otilonium bromide (Baldi 1982; Baldi 1991; Battaglia 1998; Evangelista 1999; Glende 2002)
- Pinaverium bromide (Awad 1997; Corazza 1983; Delmont 1981; Dubarry 1977; Galeone 1986; Levy 1977; Lu 2000; Virat 1987)
- Prifinium bromide (Piai 1979; Sasaki 1985)
- Propinox (Pulpeiro 2000)
- Rociverine (Ghidini 1986)
- Secoverine (Ehsanullah 1985)
- Syntropium bromide (Galeone 1990)
- Trimebutine (Dumitrascu 2000; Ghidini 1986; Luttecke 1980; Moshal 1979; Schaffstein 1990; Schang 1993)
- Zamifenacin (Houghton 1997)
There were 23 included studies remaining. Nine were conducted in the UK (Dew 1984; Evans 1998; Flavell Matts 1967; Gilbody 2000; GP research group; Mitchell 2002; Nash 1986; Prout 1983; Ritchie 1979); eight in the rest of Europe, one in the USA, three in Australia and New Zealand, one in India, and one in Taiwan.
Generally, we excluded reports of studies if they were not in English, however, some studies translated and included in the Cochrane review were exceptions to this (Berthelot 1981; Czalbert 1990; Lech 1988; Schafer 1990). We had some reservations about doing this because of a difference of opinion with the Cochrane review on the eligibility of one of the English-language studies (Fielding 1980) due to the absence of reporting randomisation, but the study was included in the Cochrane review. However, we decided to include all the non-English language studies in the Cochrane review on the basis of trust. There was only one paper (Czalbert 1990) for which we were unable to verify that the patients were randomised to treatments, and we decided to include this and carry out a sensitivity analysis.
The majority of studies (17/23) had fewer than 100 patients, with six having 20 or fewer in the intervention arm (Carling 1989; Czalbert 1990; Evans 1998; Lech 1988; Ritchie 1979; Tasman-Jones 1973). Six studies had more than 100 patients in total and one had 712 patients (Schafer 1990). Some of the studies were of crossover design, so fewer patients are required to achieve adequate power.
Study Design
Setting: The majority of studies took place in secondary care; two studies were in primary care (Gilbody 2000; GP research group); five studies did not report the setting or it was not possible to determine it because the report was not in English (Berthelot 1981; Czalbert 1990; Evans 1998; Flavell Matts 1967; Schafer 1990).
There were ten crossover studies (Dew 1984; Evans 1998; Flavell Matts 1967; GP research group; Hennessy 1975; Lawson 1988; Nash 1986; Prout 1983; Tasman-Jones 1973; van Outryve 1995) in which participants were allocated to receive both the intervention and control treatments during the course of the study, in a random order. All of these studies had either no ‘washout period’ or it was not reported, in which case this was assumed to be none. As the GDG had specified a washout period of one week minimum, all these studies were eliminated from the analysis. One additional crossover study (Carling 1989) reported first period results for the global improvement of symptoms outcome, so these data were combined with those from parallel design studies.
The remaining 12 studies had a parallel design (Berthelot 1981; Czalbert 1990; Gilbody 2000; Inauen 1994; Kruis 1986; Lech 1988; Liu 1997; Mitchell 2002; Nigam 1984; Page 1981; Richie 1979; Schafer 1990).
Thirteen studies were therefore included in the analysis (12 parallel and one crossover trial, first period only).
Two studies had more than two arms: Schafer (1990) compared hyoscine plus paracetamol and hyoscine alone with placebo; Carling (1989) compared peppermint oil with hyoscamine and placebo; Ritchie (1979) compared hyoscine with placebo, and hyoscine+lorazepam with lorazepam alone. There were thus 17 comparisons in the antispasmodics review.
The rest of the description of studies will focus on these studies/comparisons.
Population
The definition of IBS varied between studies: one used the Rome I criteria (Mitchell 2002); one used the Rome II criteria (Gilbody 2000); three met criteria defined by the authors that were similar to the above (Inauen 1994; Nigam 1984; Page 1981). In seven comparisons, the authors stated that the patients had IBS, with no further explanation (Berthelot 1981; Czalbert 1990; Liu 1997; 2 x Ritchie 1979; 2 x Schafer 1990). The remaining five comparisons (3 x Carling 1989; Kruis 1986; Lech 1988) did not use a formal definition but described a range of symptoms consistent with IBS.
Most studies included a combination of IBS types, one study specified constipation (Page 1981); one study (Carling 1989) included only patients with IBS-C and IBS-A; and eight were unclear (Berthelot 1981; Czalbert 1990; Gilbody 2000; Lech 1988; Liu 1997; Nigam 1984; 2 x Ritchie 1979).
None of the studies stated that any participants had IBS as result of gastrointestinal infection.
The majority of studies did not state the number of participants with bloating. Four studies had some patients with bloating (Czalbert 1990; Inauen 1994; Kruis 1986; Liu 1997). Three comparisons (3 x Carling 1989) stated that all patients had bloating.
Most of the studies did not describe symptom severity. Two studies stated that participants had symptoms of mixed severity (Inauen 1994; Mitchell 2002).
The age range of participants across studies was 16 to 79 years, with the mean age (where given) ranging from 34.5 to 48 years (Czalbert 1990). None of the studies particularly identified elderly participants.
Most studies had more women than men; two studies had more men than women (Liu 1997; Nigam 1984).
Interventions
The studies varied in the type of antispasmodic used:
- Eight comparisons used anti-muscarinics:
- One dicyclomine bromide (Page 1981)
- Two hyoscamine (2 x Carling 1989)
- One hyoscine plus paracetamol (Schafer 1990)
- One hyoscine plus diazepam (Ritchie 1979).
- Nine had direct-action smooth muscle relaxants:
- One alverine (Mitchell 2002)
- Four mebeverine (Berthelot 1981; Kruis 1986; Gilbody 2000; Inauen 1994 (the last two were also modified release)
None of the included studies used antispasmodics for short-term symptom relief. The duration ranged from two weeks to 16 weeks (Kruis 1986). The most common durations were 12 weeks (four studies), four weeks (four studies) and two weeks (four studies). Studies of less than 4 weeks duration were considered in sensitivity analyses because the GDG preferred a minimum of four weeks duration. This meant the following: two weeks – four studies (3 x Carling 1989; Page 1981); three weeks – one study (Inauen 1994).
Comparisons
The included studies covered the following comparisons (we have indicated with an asterisk the studies with less than 4 weeks duration):
- 13 comparisons of antispasmodics versus placebo:
- Seven gave direct-action smooth muscle relaxants (Berthelot 1981; Carling 1989*; Czalbert 1990; Kruis 1986; Lech 1988; Liu 1997; Mitchell 2002)
- One study (Ritchie 1979) compared antispasmodic plus diazepam versus diazepam alone
- One study (Schafer 1990) compared antispasmodic plus paracetamol versus paracetamol alone
- One study compared different classes of antispasmodic:
- One compared an anti-muscarinic with a smooth muscle relaxant (Carling 1989*)
- Two studies compared different types of antispasmodic in the same class (smooth muscle relaxant):
- Two studies (Gilbody 2000; Inauen 1994*) compared mebeverine modified release (200 g bid) with mebeverine conventional (135g tid).
Methodological Quality
The results of the quality assessment for included trials are shown in Appendix D.
The method of randomisation was reported in none of the studies. Allocation concealment was reported in one study (Ritchie 1979), which reported an adequate method, in which the drugs were issued in random order by the pharmacist. All but two of the studies reported that the patients were blinded to the interventions. One stated that the patients were not blinded (Inauen 1994). One study (Czalbert 1990) was unclear about patient blinding. Only one study (Mitchell 2002) described an a-priori power calculation. Several studies included in the review demonstrated baseline comparability of the groups, but 11 did not give baseline characteristics or were in non-English languages (Berthelot 1981; Lech 1988; Nigam 1984; Ritchie 1979; Schafer 1990). One study was not comparable at baseline (Liu 1997) for the severity of stool frequency (more severe for Colpermin). Three studies reported no withdrawals (Czalbert 1990; Nigam 1984; Ritchie 1979). One study (Page 1981) reported that more than 20% of patients in at least one arm (or overall) were not analysed or were lost to follow-up (attrition bias): 33% in the intervention group and 39% in the placebo group.
The GDG preferred a minimum intervention period of four weeks as this was felt to be clinically significant relative to potential effect. This meant the following were treated with caution: two weeks: Carling (1989); Inauen (1994); Page (1981).
The risk of bias was assessed for each included study and no studies were excluded from the analysis. Four studies were assessed as being at higher risk of bias: Page (1981) (attrition bias and duration less than four weeks); Inauen (1994) (patients not blinded and duration less than four weeks); Liu 1997 (lack of baseline comparability), and; Carling (1989) (duration less than 4 weeks). All these studies were treated with caution and sensitivity analyses were carried out. This was also done for Czalbert as discussed earlier.
RESULTS
A. Antispasmodics versus Placebo
There were 12 studies included in the analysis that compared antispasmodics with placebo, and a further two studies that compared antispasmodics + another intervention versus that other intervention alone. The GDG decided that it was inappropriate to combine the two types of comparison, even though each could be considered to be a comparison of antispasmodics versus placebo. The GDG’s view was that the drugs could interact and their effects would not simply be additive.
One of the studies was in patients with constipation-predominant IBS (Page 1981); eight did not specify the type of IBS and the remainder had patients of mixed IBS-type. Therefore the studies were not stratified by IBS type. Similarly, there was too little information to separate by severity, post-infective cause or bloating status. Only one study (Mitchell 2002) reported using established criteria to diagnose IBS (Rome I).
Where outcomes were measured at different times during the study, we took the end-study results unless there were significant numbers of withdrawals or problems with compliance. Therefore, for the Kruis (1986) study we took the values at four weeks.
1. Global symptoms
a. Number of patients with improvement in global symptoms
Eight studies with 731 patients reported this outcome for the comparison antispasmodics versus placebo. As described in the general section, patient assessments of improvement were combined with symptom score related methods (unlike the Cochrane review). Overall the relative risk was 1.32 (95%CI 1.18, 1.48), i.e. statistically significant, favouring antispasmodics. There was some heterogeneity, but it was not significant (p=0.09; I2=43%). The sensitivity analysis without Page (1981) made little difference to the summary statistics.
The full meta-analysis corresponded to a number needed to treat (NNT) of 6 (95%CI 5, 10), for a placebo group rate of 0 to 71%. This meant that clinicians needed to treat 6 patients to get additional benefit in relief of symptoms for one patient. Typically this is viewed as a low NNT.

Figure 1
Subgroup analysis into anti-muscarinic agents and direct-acting smooth muscle relaxants, for all antispasmodics-placebo comparisons (Figure 2) suggested there was little difference between classes of antispasmodics, although there was some heterogeneity (I2=57%, p=0.08) for the anti-muscarinics group. The RR for the random effects model was 1.51 (95%CI 1.00, 2.28), i.e. this result was sensitive to the method of analysis.

We noted that three of these studies were in hyoscine and the remaining one (Page 1981) was in dicyclomine bromide. Page (1981) had over 30% missing data and was only two weeks in duration. In its absence there was little difference to the fixed effects result, but a substantial difference to the random effects model.
b. Global symptom score (mean)
This outcome was recorded by one study (Carling 1989), which compared the change in symptom score (before and after intervention) for peppermint oil and hyoscamine (atropine) compared with placebo. This was a crossover study that reported first period results. The study reported p values for the difference:
- Peppermint oil versus placebo: mean difference for change score was −11.8 (on a scale of 105 for a week); p=0.063 (i.e. statistically significant).
- Atropine versus placebo: mean difference for change score was −1.0 (on a scale of 105 for a week); p=0.46 (i.e. not statistically significant).
2. Individual symptoms
a. Pain
The following studies measured pain:
- Number of patients with no pain: one study (Liu 1997)
- Number of patients with less pain: four studies (Lech 1988; Liu 1997; Mitchell 2002; Page 1981 (physician assessed)). The Kruis (1986) study was not included because of poor compliance – 4 week results were not reported for this outcome.
- Pain score (change): one study (Berthelot 1981).
Figure 3 shows the number of patients with less pain. There was a statistically significant increase in the number with less pain, favouring antispasmodics. However, there was also significant heterogeneity within the smooth muscle relaxants group (p=0.07, I2=63%). This was possibly a drug specific effect: Lech (1988) and Liu (1997) gave peppermint oil and Mitchell (2002) gave alverine citrate. However, the duration of Mitchell (2002) was also longer (12 weeks versus 4 weeks).

A sensitivity analysis without Mitchell (2002) shows that this was responsible for the heterogeneity, but it was not clear why. It was interesting to note that this study was the only one that carried out a power calculation.

Figure 4Sensitivity analysis
The one study (Berthelot 1981) recording a pain score showed a statistically significant decrease in pain score, favouring the antispasmodic (figure 5). The mean difference was −0.56 (95%CI −1.03, −0.09) on a scale of 1 to 4.

b. Bloating
Only two studies (Liu 1997 and Mitchell 2002) reported the number of patients with less bloating (Figure 6). Meta-analysis showed a high degree of heterogeneity between studies (p=0.0002, I2=93%). It was not clear why these studies were so different, but see the discussion above regarding the pain outcome.

c. Bowel habits
i. Number of patients with improved bowel habits
One study (Page 1981), with 71 patients and attrition bias, recorded the number of patients with improved bowel habits (figure 7). There was a statistically significant improvement with the antispasmodic.
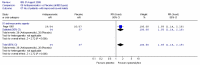
ii. Stool score
One study reported a stool score (Berthelot 1981) on a scale of 1–4. There was a statistically significant difference between antispasmodic and placebo, favouring the former.

Figure 8
3. Adverse effects
Three studies (four comparisons) reported the number of patients with adverse effects. These were grouped by antispasmodic. In all cases there were wide confidence intervals, but it appeared that there were significantly more side effects for both atropine and dicyclomine bromide than placebo (Figure 9). In particular, the statistically significant effect of atropine was manifested as dry mouth and blurred vision, but the confidence intervals were very wide, as demonstrated in figure 10.


Figure 10
Specific side effects.
B. Antispasmodic type 1 versus Antispasmodic type 2
One study compared different types of antispasmodic: Carling (1989) compared hyoscamine (atropine) with peppermint oil (smooth muscle relaxant).
1. Global outcomes
a. Global symptom score
The study did not record standard deviations or p-values.
2. Adverse effects
The study showed a statistically significant increase in side effects for atropine compared with peppermint oil, although the confidence interval was wide.

Figure 11
C. Comparison of antispasmodics in the same class
Two studies (Gilbody 2000; Inauen 1994) compared modified release mebeverine twice daily (total 400 mg) with conventional mebeverine three times daily (total 405 mg). The two studies had different durations: Gilbody (2000) had duration of eight weeks, and; Inauen (1994) had duration of three weeks. Gilbody (2000) was also carried out in general practice.
1. Global improvement of symptoms
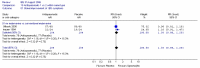
2. Adverse effects
a. Pain
One study in general practice (Gilbody 2000) showed little difference between the interventions for the number of patients with adverse effects, although it was noted that the number of adverse effects was high in both groups and the study said this included symptoms associated with IBS (pain and diarrhoea).

Figure 13
Adverse Effects
An adverse effects review has been carried out and is reported in section 8.5.1. There were six included RCTs in the review of adverse effects of antispasmodics (Grillage 1990; Gilbody 2000; Schaffstein 1990; Mitchell 2002; Liu 1997; Van Outryve 1995). The interventions and comparators were extremely varied, as was the reporting of adverse effects outcomes. In view of this, no meta-analysis was performed. Dry mouth, drowsiness, dizziness and constipation were the most common complaints reported amongst people taking antispasmodics.
One of the limitations of the adverse effects review was that many of the adverse outcomes of interest were very similar to the symptoms of the IBS itself. For instance, antispasmodics were associated with drowsiness and constipation, both of which are commonly seen in people with untreated IBS. This made it difficult for the investigators to differentiate between the progress of the condition and the harmful effects of the drug.
ECONOMIC LITERATURE FOR ANTISPASMODICS
No relevant health economic analyses were identified on the cost-effectiveness of antispasmodic therapy in the treatment of IBS.
COST-EFFECTIVENESS ANALYSIS FOR ANTISPASMODICS
This section describes the health economic analysis undertaken to inform recommendations on the use of antispasmodics as a long-term maintenance therapy in IBS. The general methods used in the economic analysis for all management interventions are described in detail in Chapter 5 and the model inputs and assumptions relevant to this particular intervention are described below.
- The following interventions were treated as a class of interventions with the same clinical effectiveness as there was insufficient evidence to demonstrate a significant difference in effectiveness between them; hyoscine, mebeverine (standard and slow release), peppermint oil, dicycloverine (dicylcomine), alverine.
- There was insufficient evidence to demonstrate that atropine (hyoscamine) was more effective than placebo. Therefore, the cost-effectiveness of atropine was not estimated.
- The studies included in the clinical effectiveness review did not stratify results by IBS subtype, so it was not possible to estimate the effectiveness for each of the subtypes separately. Therefore, it was assumed that antispasmodics are equally effective in all subtypes.
Modelled response rates
In the basecase scenario the response rate of 45% in the no treatment arm is taken from the Mearin (2004) cohort study. This represents the group of patients for whom we would expect an improvement in symptoms without any specific intervention. The RR of response for antispasmodics versus placebo is 1.32; therefore the response rate in the intervention arm is 59% (=45% × 1.32), giving an absolute difference in response between the intervention and no treatment arms of 14% (=59%–45%) during the 1st month. In the basecase scenario the response rate for the subsequent interventions is assumed to be equal to the response rate for the first intervention. Therefore an additional 14% of those who do not respond to the first intervention achieve a therapeutic response to the second intervention, increasing the overall response rate to 65% (=59%+14%*[1–59%]) after the second month. The response rate over time for the basecase is given in Figure 14.
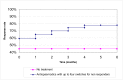
Figure 14
Modelled response rate for antispasmodic therapy, when allowing up to 4 switches of antispasmodic therapy, and no treatment.
We have also considered an alternative scenario in which no patient in the comparator arm achieves an improvement in symptoms but there is still a 14% chance of response for the intervention arm.
Table 1Intervention specific parameters – antispasmodics
| Description | Value | Evidence | |
|---|---|---|---|
| RR of response for intervention vs placebo | 1.32 (1.18 – 1.48) | Meta-analysis of RCT evidence for improvement in global symptoms | |
| Maximum number of switches considered | 4 | Limited by number of effective interventions | |
| Drug costs | |||
| Intervention | Dose per day | Cost per month* (assuming lowest cost preparation) | |
| Hyoscine | 30mg | £4.22 | |
| Mebeverine | 400mg | £6.76 | |
| Peppermint oil | 0.6ml | £7.65 | |
| Alverine | 360mg | £20.99 | |
| Dicyclomine hydrochloride | 160mg | £24.54 | |
- *
British National Formulary (Joint Formulary Committee 2007)
Results
Table 2 gives the incremental cost-effectiveness for several treatment pathways with different numbers of therapy switches included. These results demonstrate that although a treatment pathway which allows up to 4 switches had a cost per QALY under £20,000 compared to no treatment, the incremental cost-effectiveness compared to a pathway which allows up to 3 switches was greater than £20,000. Therefore, at a willingness to pay threshold of £20,000 per QALY, it would only be cost-effective to allow up to 3 switches as the additional switch would not provide sufficient additional benefit.
Table 2
Incremental cost-effectiveness of allowing subsequent switches in antispasmodic therapy.
These results are an estimate of the cost-effectiveness over the first 6 months after the initiation of antispasmodic therapy. The cost per QALY for continuing antispasmodic therapy beyond 6 months is lower than the cost per QALY during the initial 6 months provided that treatment is reviewed every 6 months and discontinued in patients who no longer experience a therapeutic benefit, either due to lack of effectiveness or a change in their symptom profile.
The probabilistic sensitivity analysis provided an estimate of the uncertainty in the cost per QALY estimate due to uncertainty in the efficacy estimate, the probability of response in the no treatment arm and the utility gain. The CEAC in Figure 15 shows the uncertainty surrounding the relative cost-effectiveness of allowing each additional switch in therapy. For example, the first curve on the left of Figure 15 shows that it is highly likely that using a first antispasmodic therapy would be cost-effective compared to no treatment as the probability of the cost per QALY being under a £20K threshold is over 90%. The second curve from the left is very close to the first and it shows that it is highly likely that allowing patients to switch once to an alternative antispasmodic therapy would be cost-effective compared to no further antispasmodic therapy for those who do not respond to the first. Similarly, the second switch is also highly likely to be cost-effective as it has a 91% probability of being under £20K. However, once the third switch of therapy is considered, it is only fairly likely to be cost-effective as there is a 53% likelihood that the true cost per QALY is under £20K and a 79% likelihood that it is under £30K. Providing four switches is fairly unlikely to be cost-effective compared to 3 switches as the cost per QALY has a 23% probability of being under £20K and a 51% probability of being under £30K. However, it should be noted that these estimates only consider the uncertainty in cost-effectiveness due to the accuracy of several input parameters and they do not reflect general uncertainty around the assumptions made in the model. The uncertainty from these assumptions was explored in the univariate sensitivity analysis.
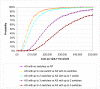
Figure 15
Cost-effectiveness acceptability curves for antispasmodics (AS) compared to no treatment (NT) and for each additional switch of antispasmodic for non responders (AS).
Univariate sensitivity results for up to three switches
The results in Table 3 show that initiating treatment with an antispasmodic and allowing up to three switches in therapy for non-responders had a cost per QALY of £7,952 compared to no treatment. When the response rate in the no treatment arm was taken from the average response rate in the RCTs of pharmacological interventions (47%), rather than from the cohort study (45%), the cost per QALY was very similar at £7,539. Maintaining the 14% difference in response between the two arms but reducing the response rate in the no treatment arm from 45% to zero marginally decreased the cost per QALY to £7,772. Reducing the response rate for subsequent antispasmodics, in patients who have not responded to initial therapy, by 50% significantly increased the cost per QALY for each subsequent switch of therapy, such that only 2 rather than 3 switches of therapy could be provided for a cost per QALY under £20,000. However, the cost per QALY for 3 switches compared to no treatment was only moderately increased to £10,003 per QALY and even when the response rate for subsequent therapy was set to zero, the cost per QALY for antispasmodic therapy with up to 3 switches remained under £20,000 when compared to no treatment.
Table 3
Sensitivity results for antispasmodic therapy with up to 3 switches compared to no treatment for 100 patients with IBS (all subtypes).
We carried out a threshold analysis to determine whether antispasmodic therapy would still be cost-effective for lower gains in health related quality of life. In the basecase it was assumed that patients who respond to therapy accumulate 0.071 QALYs more per annum than patients who do not respond. For comparison, a gain of 0.135 QALYs would represent a complete remission of IBS symptoms. When the QALY gain associated with a response to therapy was reduced to 0.028 QALYs, the cost per QALY of providing antispasmodic therapy with up to 3 switches was estimated to be above £20,000 per QALY compared to no treatment.
If a patient takes a therapy on an as needed basis, it would be reasonable to assume that they take the therapy on days when their quality of life is significantly affected by their IBS symptoms but not on days when their symptoms are less severe. It has therefore been assumed in the model that they only accrue QALY benefits and drug costs on the days they take the therapy. However, it is still necessary to assess all patients for response after 1 month of initiating therapy. This means that it would be less cost-effective to initiate therapy in patients who use the therapy on fewer days, as the monitoring costs are just as high but the benefits are lower. This is shown by the estimated cost per QALY of £20,578 for patients who take the therapy on 25% on days. However, a more detailed look at the results for these patients (data not tabled) shows that up to 1 switch of therapy could be provided for a cost per QALY of £19,414 with a 45% likelihood of being under £20,000 per QALY and a 73% likelihood of being under £30K per QALY. So, cost-effective treatment strategies may be available for patients who do not experience severe symptoms as frequently.
If a patient also takes another medication (such as a laxative or anti-motility agent), then these medications can be reviewed at the same time, so it may be cost-effective to provide both therapies. For example, if laxatives are prescribed with the antispasmodic and both used on 25% of days then allowing up to 2 switches of both treatments was estimated to be cost-effective with a cost per QALY of £10,107 compared to no treatment a cost per QALY of £17,393 compared to 1 switch.
GDG DISCUSSION
The GDG consensus was that antispasmodics should be used as first line therapy alongside dietary and lifestyle advice for people with IBS, particularly those with pain occurring as spasm. Antispasmodics should be taken as needed.
EVIDENCE STATEMENTS
For this review, the evidence was assessed using the GRADE process and tables are shown in Appendix F. The following evidence statements are derived from the GRADE tables.
- There is a fair amount of good quality evidence showing significant improvement in symptoms for antispasmodics, in general, and smooth muscle relaxants, in particular, when compared to placebo.
- There is a fair amount of evidence showing significant global improvement in symptoms for antimuscarinic agents compared with placebo.
- Sub-group analysis suggests there is little difference in the effect of antimuscarinic agents and smooth muscle relaxants for global improvement of symptoms.
- There is a moderate amount of good quality evidence showing a significant reduction in pain and an improvement in bowel habit for antispasmodics compared with placebo.
- There is a moderate amount of good quality evidence showing that conventional and modified release mebeverine were equally effective.
ADVERSE EFFECTS EVIDENCE STATEMENT
There is limited evidence that antispasmodics are associated with dry mouth, dizziness and drowsiness.
HEALTH ECONOMIC STATEMENT
Evidence from a decision analytic model showed that antispasmodics (hyoscine, mebeverine, peppermint oil, dicycloverine, alverine) are cost-effective for long-term maintenance use in individuals with IBS. The cost-effectiveness estimate is based on a clinical pathway in which response is assessed after one month and non-responders are switched to an alternative antispasmodic with the lower cost antispasmodics used before higher cost antispasmodics. The analysis assumes that the response to each subsequent therapy is independent of the response to previous antispasmodics. Trying a further antispasmodic is unlikely to be cost-effective in individuals who have not responded to four previous antispasmodics. The cost-effectiveness analysis assumes that treatment is reviewed every 6 months to establish whether antispasmodic therapy is still relevant to the individual’s symptom profile.
EVIDENCE TO RECOMMENDATIONS
The GDG took into consideration the clinical and cost effective evidence. They noted the limited evidence about adverse effects, but did not consider this to be a significant factor in practice. The GDG wished to encourage primary care clinicians to give antispasmodics as a first line therapy alongside dietary and lifestyle advice.
RECOMMENDATION
Healthcare professionals should consider prescribing antispasmodic agents for people with IBS. These should be taken as required, alongside dietary and lifestyle advice.
8.4. Antidepressants
SELECTION CRITERIA
The selection criteria described in the general methodology section were used, but some criteria specific to the antidepressants review are reported below.
Types of patients
For this review, patients were required to have IBS and not to have inflammatory bowel disease or major psychiatric disorders.
Types of studies
Studies that investigated drugs not listed in the BNF were excluded. These included: Amineptine; Amoxapine; Desipramine, and; Pirenzepine.
The GDG decided that crossover studies were acceptable and that the washout period for this review should be at least one week. Trials with shorter washout periods were not included in the analysis.
Types of intervention
Studies included the following interventions:
Tricyclics and related antidepressants:
- Amitriptyline (Triptafen®, Triptafen-M® (with perphenazine)
- Clomipramine (Anafranil®)
- Dosulephin (Prothiaden®)
- Doxepin (Sinepin®)
- Imipramine
- Lofepramine (Feprapax®, Lomont®, Gamanil®)
- Nortriptyline (Allegron®)
- Trimipramine(Surmontil®)
- Mianserin
- Trazodone (Molipaxin®).
Selective serotonin re-uptake inhibitors (SSRIs):
- Citalopram (Cipramil®)
- Escitalopram (Cipralex®)
- Fluoxetine (Prozac®)
- Fluvoxamine (Faverin®)
- Paroxetine (Seroxat®)
- Sertraline (Lustral®).
Monoamine oxidase inhibitors (MAOIs):
- Phenelzine (Nardil®)
- Isocarboxazid
- Tranylcypromine.
Reversible MAOIs:
- Moclobemide (Manerix®).
Others:
- Duloxetine (Cymbalta®)
- Flupentixol (Fluanxol®)
- Mirtazapine (Zispin Soltab®)
- Reboxetine (Edronax®)
- L-Tryptophan (Optimax®).
The following comparisons were included:
- Antidepressant versus placebo (or nothing)
- Antidepressant type 1 versus Antidepressant type 2
- Antidepressant dose 1 versus Antidepressant dose 2
- Antidepressant versus other interventions.
NB: In spite of the large placebo effect associated with IBS, comparisons with no treatment were included.
The antidepressants review was concerned with medium and longer term symptom relief. Medium term treatment was defined as three months and long term as between six months and one year.
Subgroup analyses
We planned to carry out subgroup analyses by type of antidepressant; dose; mode of delivery, and; duration of intervention.
Search strategy for identification of studies
Searches were performed on the following core databases: MEDLINE; EMBASE; CINAHL, and; The Cochrane Library (1966 to current day with guidance from the GDG). Additional databases were not searched for this review. The search strategies are listed in Appendix B.
The search strategy identified 1458 studies. The titles and abstracts of these studies were assessed. Thirty were identified to be potentially relevant to the review and these papers were retrieved in full. The reference lists for each of the retrieved studies were inspected for further potential papers, but none were identified. The 17 excluded studies are listed in the Appendix, along with reasons for exclusion. The remaining 13 studies were included (Boerner 1988; Creed 2003; Kuiken 2003; Myren 1982; Myren 1984; Rajagopalan 1998; Schrivastava 1984; Steinhart 1982; Tabas 2004; Tanum and Malt 1996; Tripathi 1983; Vij 1991; Quartero 2007). One of these studies was a Cochrane review (Quartero 2007). The Myren (1982) study did not state that it was randomised although it was double blind. Since this study was included in the Cochrane review (under the name of Block (1983), which is an identical paper in Norwegian) we included it too, but treated it with caution.
DESCRIPTION OF STUDIES INCLUDED IN THE REVIEW
One Cochrane review was identified (Quartero 2007) and this guideline review is an update, with revision to make it appropriate to the UK. This mainly involved elimination of one of the included studies (Heefner 1978) because the antidepressant, desipramine, is not in the BNF. Therefore the analysis for the guideline review was based on six studies included in the Cochrane review (Boerner 1988; Myren 1982; Myren 1984; Rajagopalan 1998; Shrivastava 1984; Vij 1991) and six additional studies (Creed 2003; Kuiken 2003; Steinhart 1982; Tabas 2004; Tanum and Malt 1996; Tripathi 1983). Tripathi (1983) and Shrivastava (1984) were two reports of a single trial, i.e. there were 11 included studies.
One study was conducted in the UK (Creed 2003); one was conducted in Germany (Boerner 1988); one in The Netherlands; three in Norway (Myren 1982; Myren 1984; Tanum and Malt 1996): three in India (Rajagopalan 1998; Tripathi 1983; Vij 1991) and the remaining two in the United States (Steinhart 1982; Tabas 2004).
Eight studies had fewer than 100 patients (Myren 1982; Boerner 1988; Kuiken 2003; Steinhart 1982; Rajagopalan 1998; Tripathi 1983; Tabas 2004; Tanum and Malt 1996). Two studies had fewer than 20 patients in the intervention arm (Kuiken 2003; Steinhart 1982). The latter was a crossover design so fewer patients were required to achieve adequate power. Creed (2003) had 257 patients and Myren (1984) had 258.
Study Design
Setting: The majority of studies took place in secondary care, one of which treated inpatients (Tripathi 1983); one was stated to take place in primary care in a later paper (Myren 1982); three included patients from both primary and secondary care settings (Boerner 1988; Myren 1984; Tabas 2004) and one study (Steinhart 1982) did not report the setting.
Funding: three studies received some funding from industry: Kuiken (2003) was sponsored by Eli Lilly (manufacturers of fluoxetine); Tripathi (1983) received the study medication and placebo from May & Baker India Ltd. (manufacturers of trimipramine); Steinhart (1982) received the study medication and placebo from Merck Sharp & Dohme. Six studies (Myren 1982; Myren 1984; Rajagopalan 1998; Vij 1991; Boerner 1988) did not state source of funding. The remaining studies were funded by non-industry sources (Tabas 2004; Tanum and Malt 1996; Creed 2003).
Population
The age range of patients across the IBS studies was 13 to 75 years, with the mean age, where given, ranging from 35 to 41 years. The study with a lower age of 13 was Tripathi (1983); in this study the range was 13 to 60 years, with a mean of 37. The GDG did not consider this level of children to be important. No study particularly identified elderly patients.
Five studies had more women than men (Creed 2003; Myren 1982; Steinhart 1982; Tabas 2004; Tanum and Malt 1996); three studies had about the same number of men and women (Kuiken 2003; Rajagopalan 1998; Myren 1984) and two studies did not give the proportions of patients by gender (Boerner 1988; Tripathi 1983).
Six studies identified the type of IBS as mixed (Boerner 1988; Creed 2003; Kuiken 2003; Steinhart 1982; Tabas 2004; Vij 1991). The remaining studies gave no information regarding type of IBS. In Tanum and Malt (1996) only 60% of the patients had IBS; the rest had non ulcer dyspepsia (NUD). The authors reported that there was no significant difference in response between IBS and NUD patients, although there was a trend towards a slightly better response in the NUD patients.
Two studies stated that the patients had severe IBS (Creed 2003; Steinhart 1981); one study had mixed severity patients (Vij 1991). Creed (2003) was stratified by pain level before randomisation. Tabas (2004) also selected patients who were non-responders to placebo.
Four studies reported the inclusion of refractory IBS patients: Creed (2003) included patients that had failed to respond to usual treatment and had a median duration of IBS of eight years; Kuiken (2003) stated that the patients had all been treated unsuccessfully previously. Steinhart (1981) reported a mean duration of IBS of 5 years and stated that all patients had received antispasmodics previously; Tabas (2004) included patients who had failed to respond to a high fibre diet.
Three studies reported a long duration of IBS: Tanum and Malt (1996) had a mean duration of symptoms of about 8 years; Rajagopalan (1998) had a mean duration of about 4 years; Kuiken (2003) had a mean duration of symptoms of 5.9 years.
One study implied that some of the patients did not have refractory IBS: in Myren (1984), 45 to 61% were not taking other drugs before the study started, and the other studies did not report previous treatments.
Five studies stated that the patients had some depression: Tabas (2004) reported that 27/81 (33%) had a score greater than 10 on the Beck Depression Inventory (although major psychiatric illnesses were excluded); Creed (2003) reported that 47% had anxiety or depression; Steinhart (1981) stated that 57% had depression and 79% anxiety; Vij (1991) had 57% with psychiatric co-morbidities; Boerner (1988) reported that some patients had depression (mid point on the Hamilton depression scale). Kuiken (2003) excluded patients with depression.
One study stated that all the patients had anxiety (Tripathi 1983). Three studies reported that the patients did not have psychiatric disorders: Rajagopalan (1998) stated that patients had no major medical or psychiatric illnesses; Tanum and Malt (1986) excluded patients with schizophrenia, anxiety or depression, and; Myren (1982) and Myren (1984) both showed low scores on depression and anxiety scales.
Interventions
The interventions included:
- Tricyclic Amitriptyline up to 75mg for 3 months (Rajagopalan 1998) and 50mg for 1 month (Steinhart 1982) – c.f. BNF levels for depression treatment: initially 75mg, increasing to 150 to 200mg
- Tricyclic Trimipramine 30 to 50mg for 4 to 6 weeks in three studies (Myren 1982; Myren 1984; Tripathi 1983) – c.f. BNF levels for depression: initially 50–75mg, then 150 to 300mg
- Tricyclic Doxepin 50mg for 8 weeks (Boerner 1988) and 75mg for 6 weeks (Vij 1991) – c.f. BNF levels for depression: initially 75mg, then 30 to 300mg
- Tricyclic-related Mianserin 30 mg initially, then up to 120mg for weeks 2 to 7, then tapered in week 8 – c.f. BNF levels for depression: 30 to 40mg initially; usual dose 30 to 90mg
- SSRI Paroxetine 20mg per day for 3 months (Creed 2003) and up to 40mg for 3 months; 23% 10mg; 43% 20mg; 33% 40mg (Tabas 2004) – c.f. BNF levels for depression: initially 20mg, then up to 50mg
- SSRI Fluoxetine 20mg per day for 6 weeks – c.f. BNF levels for major depression: 20mg once daily increased after 3 weeks if necessary, usual dose 20 to 60mg.
Comparisons
The majority of comparisons were of antidepressants versus placebo. One study (Creed 2003) compared antidepressants with usual care. One study compared different doses of antidepressants (Myren 1984).
Antidepressant versus placebo
- Tricyclics versus placebo:
- Trimepramine (Myren 1982; Myren 1984; Tripathi 1983)
- Amitriptyline (Rajagopalan 1998; Steinhart 1982)
- Doxepin (Boerner 1988; Vij 1991)
- Mianserin (Tanum and Malt 1996).
- SSRI versus placebo:
- Paroxetine (Tabas 2004) included high fibre diet (>25g daily) in both the treatment and placebo groups (NB. these patients had already been identified as non-responders to high fibre)
- Fluoxetine (Kuiken 2003).
Antidepressant versus usual care
- SSRI versus usual care:
- Paroxetine (Creed 2003) versus ‘routine care’ by gastroenterologist and GP including antispasmodics, laxatives, antidiarrhoeal medication or additional analgesics.
Antidepressant versus psychotherapy (this is covered in the psychotherapy review)
- Paroxetine versus psychotherapy (Creed 2003).
It was decided to combine the SSRI studies with comparators of placebo and usual care using subgroup analyses.
METHODOLOGICAL QUALITY
The results of the quality assessment for included trials are shown in Appendix D.
The method of randomisation was adequate in four of the studies: three used computer generated random numbers (Creed 2003; Kuiken 2003; Tabas 2004) and the other used a random number table (Vij 1991). Myren (1982) did not state that the study was randomised, but stated that the study was double blinded; we included this study because it was stated to be an RCT in the Cochrane review. The remaining studies did not state the method of randomisation. Allocation concealment was reported in four studies (Creed 2003; Myren 1982; Tabas 2004). Creed (2003) and Kuiken (2003) reported that an independent third party carried out the randomisation; Tabas (2004) reported that identical capsules were sealed in sequentially numbered identical boxes.
The majority of studies reported that the patients were blinded to the interventions, with the exception of Creed (2003), in which blinding was not possible due to the nature of the comparisons. However, the outcome assessors were blinded in this study.
Three studies reported a sample size calculation (Creed 2003; Kuiken 2003; Tabas 2004), but Kuiken (2003) was powered for a different primary outcome (rectal sensitivity) and was underpowered for symptoms. The remaining studies gave no details of an a priori sample size calculation (Boerner 1988; Myren 1982; Myren 1984; Rajagopalan 1998; Tripathi 1983; Steinhart 1982; Tanum and Malt 1996; Vij 1991).
The majority of studies included in the review demonstrated baseline comparability of the groups, apart from Myren (1982) which was not comparable at baseline for vomiting, but levels of this were low in both groups (0.5 and 0.1 on 10cm VAS), and; Rajagopalan (1998) which was not comparable on stool type, with the antidepressant group having looser stools. In two studies there were no details of baseline characteristics (Tripathi 1983; Boerner 1988).
Only one study had total missing data of more than 20% (Rajagopalan 1998), comprising 9/20 in each group (45%). Those who dropped out had a significantly shorter duration of symptoms at recruitment, but otherwise there was no difference between completers and dropouts. Two studies had no missing data (Tripathi 1983; Myren 1982). Four reported missing data of less than 20% (Kuiken 2003; Tanum and Malt 1996; Tabas 2004; Vij 1991). However, there were 24% missing data in each arm of Vij (1991) for the outcome of pain. Myren (1984) and Steinhart (1982) provided no information regarding the number of drop outs.
In Creed (2003) there were missing data, 16% (14/86) in the paroxetine group; 14% (12/85) psychotherapy. 0% in the routine care group did not start the trial. A further 29/86 (34%) in the paroxetine group and 14/85 (16%) in the psychotherapy arm discontinued treatment, but these patients still appear to have been followed. Overall, loss to follow-up at three months was 12/86 (14%) for paroxetine, 11/85 (13%) psychotherapy and 7/86 (8%) usual care arm. At 15 months the authors contacted more of the patients. The authors reported that there were no significant differences at baseline between those who did and did not complete the treatments. For the 3 month pain score and SF36 outcome measures respectively, the patients included in the analysis were 74 and 59 (69%) paroxetine; 74 and 58 (68%) psychotherapy and 79 and 63 (73%) usual care, but some of these patients had discontinued treatment. We decided to include the results from this study, with some reservations, especially about the paroxetine arm and about the SF36 results. The study also recorded the number of patients with an improvement in global symptoms, based on the results from 74, 74 and 80 patients respectively. The GDG decided that this outcome was more representative because patients that dropped out due to side effects would not have rated their global symptoms as improved. The follow-up period in Creed (2003) allowed the patients to have paroxetine in all arms: 42% in paroxetine group, 19% in psychotherapy and 22% in the usual care group, i.e. the follow-up period should be considered to be partly confounded. Therefore we did not report the results for the follow-up period for the comparison paroxetine versus placebo.
Overall, we considered that Rajagopalan (1998) was at high risk of bias because of the extent and nature of the missing data and the baseline differences, and we decided not to include the results from this study in the analysis. Three other studies were treated with caution: Myren (1982), which was not stated to be randomised; Creed (2003) which had missing data and non compliance for pain and SF36; and some confounding in the follow up period; and Vij (1991), which had 24% missing data in each arm for the pain outcome. We examined the latter three studies with sensitivity analyses.
RESULTS
A. Antidepressants versus placebo or usual care
1. Global symptoms
a. Number of patients with global improvement of symptoms (pain, bloating and bowel habit)
Six studies, in 434 patients, reported the number of patients with improvement in global symptoms (Boerner 1988; Creed 2003; Kuiken 2003; Tabas 2004; Myren 1982; Vij 1991). The studies were combined in a meta-analysis, but as separate subgroups by type of antidepressant. The controlled trial (Myren 1982) was included in the tricyclics subgroup and examined in a sensitivity analysis. The comparisons of paroxetine with placebo and with usual care were also considered in sensitivity analyses. ‘Usual care’ was defined as patients receiving IBS treatment that was deemed appropriate by either their gastroenterologist consultant or general practitioner.
The difference between the antidepressants and placebo was statistically significant overall and for each subgroup. Within subgroups there was no heterogeneity, but between groups there was some (I2=42%, p=0.12). The overall relative risk (RR) for the meta-analysis of 434 patients was 1.55 (95% CI 1.30, 1.84), which corresponded to a number needed to treat of 5 (95% CI 4, 7), for a control group rate of 22 to 68%.
For the tricyclic subgroup (n=180) the number with global improvement of symptoms was statistically significantly higher for the antidepressant group; RR 1.31 (95% CI 1.04, 1.64), which gave an NNT of 6 (95% CI 4, 34), for a control group rate of 22–68%. In the absence of the Myren (1982) study, which was not stated to be randomised, the RR for this group was 1.37 (95% CI 0.99 1.91), i.e. no longer significant and with some heterogeneity (I2=62%, p=0.11).
For the meta-analysis of the three SSRI studies (n=254), there was a significant difference favouring antidepressant, RR 1.80 (95% CI 1.38, 2.34), with no heterogeneity (I2=0%, p=0.48). This corresponded to an NNT of 4 (95% CI 3, 7), for a control group rate of 28–41%. In the absence of the Creed (2003) study the effect was slightly bigger (RR 1.85 (95% CI 1.17, 2.91). We decided to use the results in Figure 1 in the health economic modelling.

b. Global symptom score
One study with 28 patients reported the global symptom score, but no details of the scale were given. There was no significant difference between interventions.

Figure 2
2. Individual symptoms
a. Pain
i. Number of patients with less pain
Three studies reported this outcome. Two were tricyclics versus placebo (Vij 1991; Tanum and Malt 1996) and the other was an SSRI versus placebo (Tabas 2004). These were included as subgroups in a meta-analysis in 150 patients. Overall there was significant heterogeneity (I2=84%, p=0.004), which is attributed to the type of antidepressant.
There was a large significant difference in favour of tricyclics compared to placebo in the number of patients with reduced pain; RR 3.91 (95% CI 1.93, 7.93), with no heterogeneity (I2=0%, p=0.81), although the confidence interval was fairly wide. This corresponded to an NNT of 2 (95% CI 2, 4), for a placebo group rate of 16–18%. We noted that this analysis included Tanum and Malt (1996) which had only 60% of patients with IBS, and; Vij (1991) which had 24% missing data in each arm.
There was no significant difference between the SSRI and placebo in one study in 66 patients (Tabas 2004); RR 0.88 (95% CI 0.54, 1.45).

Figure 3
ii. Number of patients with pain
One study in 34 non-depressed patients (Kuiken 2003) reported the number of patients with ‘significant pain’. There was no statistically significant difference between interventions, although the confidence interval was fairly wide.

Figure 4
iii. Pain score
Four studies (Creed 2003; Tanum and Malt 1996; Myren 1982; Myren 1984) reported a type of pain score, all appeared to use a VAS of 100 mm or 10 cm. Myren (1982) recorded an abdominal obstruction discomfort measurement, which the GDG decided was a different outcome to pain. Myren (1984) gave means and p values only. We decided to combine the first two studies in a meta-analysis by subgroup (Creed 2003 compared an SSRI with usual care). We also noted that Tanum and Malt (1996) had only 60% of patients with IBS.

Figure 5
At the end of treatment, meta-analysis showed significant heterogeneity (I2=77%, p=0.04), with different effect sizes being found for the two studies. This may be an effect of type of antidepressant, type of comparator, severity of IBS, or it may be that Tanum and Malt (1996) overestimated the effect because there were only 60% of patients with IBS. Individually, there was a statistically significant difference for the Tanum and Malt (1996) study (tricyclic versus placebo in 60% IBS): mean difference −25.90 (95% CI −38.82, −12.98) and for the Creed (2003) study: mean difference −9.20 (95% CI −18.35, −0.05).
Boerner (1988) compared a tricyclic with placebo, and recorded the median improvement in pain on a scale of 0 to 4. There was a difference in median change score of 0.3 units, in favour of the tricyclic antidepressant, which was reported to be statistically significant at the level of p<0.05.
b. Bowel habit
i. Number of patients with improvement in bowel habit
Meta-analysis of two studies in 110 patients showed a statistically significant increase in the number of patients with improvement in bowel habit; RR 1.92 (95% CI 1.19, 3.07), with no heterogeneity between the tricyclics and SSRI subgroups (I2=0; p=0.51) corresponding to an NNT of 4 (95% CI 3, 13). The confidence interval was fairly wide. The results for individual classes of antidepressant were not statistically significant, although the antidepressant was favoured; Vij (1991) (tricyclics) had a wide confidence interval and Tabas (2004) (SSRI) was fairly wide.

Figure 6
c. Bloating
i. Number of patients with less bloating
Two studies recorded the number of patients with less bloating, one a tricyclic (Boerner 1998) and the other using an SSRI (Tabas 2004), both compared with placebo. Meta-analysis could not be carried out because Boerner (1998) reported the median.
Boerner (1988) recorded the median improvement in the feeling of fullness on a scale of 0 to 4. There was a difference in median change score of 0.23 units, in favour of the tricyclic, but this was reported to be not statistically significant.
Tabas (2004), comparing SSRI with placebo, did not demonstrate a significant effect on the number of patients with less bloating, but the confidence interval was fairly wide so this conclusion was uncertain.

Figure 7
ii. Number of patients with bloating
One study (Kuiken 2003) reported the number of patients with bloating in 34 non-depressed patients. There was no significant difference between the SSRI and placebo, although the confidence interval was fairly wide. We note that the study was sponsored by Eli Lilly (manufacturers of fluoxetine).

Figure 8
3. Mental health
a. Psychological Distress
This was measured by the SCL-90 global severity index (90 item, 5 point rating scale; range 90 to 450, high = bad). There was a small, statistically significant difference between SSRI (paroxetine) and usual care at 3 months, WMD: −0.28 (95% CI −0.43, −0.09), favouring the antidepressant.

Figure 9
b. Depression score
One study (Myren 1982, CCT) recorded depression on a VAS of 0–10 cm for tricyclics versus placebo, in which a high score indicated increased depression. There was statistically significantly less depression for the patients taking antidepressants. We noted that this study, at baseline, showed low scores on depression and anxiety scales.

Figure 10
c. Anxiety
Two studies reported anxiety; one (Myren 1982, CCT) compared tricyclics versus placebo on a VAS of 0 to 10 and the other (Tabas 2004) reported the number of patients with anxiety for the comparison SSRI with placebo. The former study, at baseline, showed low scores on depression and anxiety scales, and the latter reported that 27/81 (33%) had a score greater than 10 on the Beck Depression Inventory.
i. Anxiety score
There was no significant difference in the degree of anxiety.

Figure 11
ii. Number of patients with anxiety
The confidence interval was wide and the result was not statistically significant.

Figure 12
4. Quality of life
Two studies reported quality of life measures, both for SSRIs (Creed 2003; Tabas 2004).
a. IBS-QoL
One study (Tabas 2004) comparing SSRI and placebo examined the change compared with baseline in three components of IBS-QoL: food avoidance; work function; social function. Tabas (2004) found the following differences for SSRI versus placebo:
- Food avoidance subscale – improvement of 12.7%; p value = 0.03 (statistically significant)
- Work function subscale – improvement of 2.1%; p value = 0.08 (not significant)
- Social function subscale – improvement of 13.4%; p value = 0.76 (not significant).
b. Change in SF36 Mental component score at 3 months
There was no significant difference in the mental health quality of life SF-36 score (scale 0 to 100, high=good) at 3 months between SSRI (paroxetine) and usual care, although there were 29% who discontinued treatment and 32% loss to follow up in the paroxetine arm. This conclusion does not agree with the p values reported in Creed (2003) (p=0.007).

Figure 13
c. Change in SF36 physical health score at 3 months
The change in the physical health component also showed no significant difference at 3 months. This did not agree with the p value reported in Creed (2003) (p=0.24).

Figure 14
B. Comparison of different doses of Tricyclic antidepressant
One study (Myren 1984) compared different doses of Trimipramine in 428 participants.
1. Global symptoms
The study reported the ‘total effect of treatments’ in the opinion of the physicians. Medians with their 95% confidence intervals were reported for each group. We used this to calculate the standard deviation, which we used with the median value to compare groups. Figure 16 shows there was no significant difference between any of the doses.
Figure 16. Patient pathway for Tricyclics (TC) and SSRIs (PDF, 51K)
2. Individual symptoms
a. Number of patients with abdominal pain
The study reported a statistically significant reduction in pain in patients that were given 50 mg per day either as a single dose or in two divided doses (p < 0.01). Patients taking 35 mg in a single dose had significantly less pain than placebo (p< 0.05), and for patients taking 30 mg per day in three divided doses there was stated to be no difference between the drug and placebo (p= 0.10).

Figure 15
ADVERSE EFFECTS
The evidence on adverse effects of tricyclics and SSRIs is provided in the adverse effects review (section 8.5.2).
ECONOMIC LITERATURE FOR TRICYCLICS AND SSRIS
One relevant health economic analysis was identified on the cost-effectiveness of SSRIs in the treatment of IBS (Creed 2003), but none were identified which considered the cost-effectiveness of tricyclics. Creed (2003) was a trial based economic evaluation conducted in the UK which recruited patients from secondary and tertiary care with severe IBS. This study aimed to assess whether an SSRI (paroxetine) would be superior to usual care in reducing abdominal pain and improving quality of life and whether these improvements could be achieved at no additional cost due to treatment costs being offset by reduced health care costs. (It also included a comparison of psychotherapy with usual care). The patient population considered were secondary and tertiary care patients with severe IBS who had not responded to usual treatment. The included patients had a mean duration of IBS of 8 years. This study was considered to be relevant to patients with refractory IBS only. The SSRI intervention consisted of 20 mg of paroxetine daily for 3 months which was prescribed and monitored either by the patient’s gastroenterologist or their GP. After three months, patients in the SSRI arm returned to their GP and received usual care for one year during which time they were followed-up. In the comparator arm patients received usual care from either their gastroenterologist or their GP for the three month treatment period and the following year of follow-up. The primary outcome was abdominal pain measured on a VAS of severity with secondary outcomes considering days with pain, overall change in symptoms and HRQofL measured by the SF-36. Direct costs of health care and intervention costs were recorded.
The number of people with an improvement in global symptoms was significantly higher for SSRI at the end of treatment compared to usual care. The clinical outcomes from this trial have been summarised in detail in the clinical effectiveness review. Direct health care costs were not significantly increased for SSRI compared to usual care during the intervention period or the following year. However, the results for the follow-up period may have been partially confounded as patients in the usual care arm were also allowed SSRIs.
This study was a partial economic evaluation as it did not assess the incremental cost of any benefit achieved in the form of a cost-effectiveness ratio. The evidence provided by this study was considered to be indirect as the patients were recruited from secondary and tertiary care and costs may differ for refractory patients managed in primary care. No potential areas of significant bias were identified but the results for the follow-up year were considered to be partly confounded by the use of SSRIs in the comparator arm during follow-up. Direct health care costs were not significantly increased by SSRIs during the intervention period. However, the study was powered to detect a specific change in clinical rather than cost outcomes. As this study did not provide an estimate of the cost per QALY for SSRIs compared to usual care, it was not particularly useful in determining whether recommending SSRIs for use in the NHS would result in the efficient use of NHS resources.
COST-EFFECTIVENESS ANALYSIS FOR TRICYCLICS AND SSRIS
This section describes the health economic analysis undertaken to inform recommendations on the use of tricyclics and SSRIs as long-term maintenance therapies in IBS. The general methods used in the economic analysis for all management interventions are described in detail in Chapter 5 and the model inputs and assumptions relevant to this particular intervention are described below.
The general approach is the same as for other maintenance therapies except that the clinical pathway has been modified to allow for gradual dose increases and an additional follow-up appointment at 12 weeks. The clinical pathway was modified as follows:
- Treatment is initiated at 10 mg with dose increases of 10 mg no more frequently than every 2 weeks. Patients are encouraged to increase the dose until an effective dose has been established or side-effects become problematic up to a maximum dose of 30 mg for tricyclics and 20 mg for SSRIs. (Alternative maximum doses of 50 mg for tricyclics and 40 mg for SSRIs are considered in a sensitivity analysis).
- Follow-up appointments are required every month until an effective dose has been established and a further follow-up appointment is required 12 weeks after that point. The usual 6 monthly review is carried out as for other pharmacological interventions. A sensitivity analysis in which follow-up appointments are twice as frequent was considered in order to estimate whether monitoring patients more intensively would have a significant impact on cost-effectiveness.
- Patients who do not respond to the maximum dose are switched to an alternative tricyclic or SSRI. An incremental analysis was carried out to determine whether allowing patients who do not respond to switch to a second treatment, provides sufficient additional benefit to be cost-effective given the additional costs.
- The model assumes that the response seen in the clinical trial is gradually achieved over the range of doses considered, even if the maximum dose considered is not as high as the trial dose. For example, the RCT for tricyclics versus placebo used a range of doses from 30 to 75 mg, compared to the maximum dose of 30 mg used in the basecase model. In the economic model, we assumed that equal numbers of patients respond to the 10 mg, 20 mg and 30 mg doses with the total number of responders equal to that predicted by applying the RR from the RCTs to the response rate in the no treatment arm. Sensitivity analyses were carried out assuming all patients respond to the lowest or highest dose.
The following assumptions have been made regarding the effectiveness of tricyclics and SSRIs based on the clinical effectiveness review:
- The following tricyclics were treated as a class of interventions with the same clinical effectiveness as there was insufficient evidence to demonstrate a significant difference in effectiveness between them: trimipramine, amitriptyline, doxepin.
- There was evidence that there is no difference in the effectiveness of tricyclics for doses above 30 mg compared to 30 mg (Myren 1984), so this was the maximum dose modelled in the basecase. A more conservative assumption in which doses of 50 mg are required to achieve the effectiveness seen in the RCTs have been considered in a sensitivity analysis.
- The following SSRIs were treated as a class of interventions with the same clinical effectiveness as there was insufficient evidence to demonstrate a significant difference in effectiveness between them: fluoxetine, paroxetine. A maximum dose of 20 mg was modelled in the basecase as this was the most commonly prescribed dose in the trials, but a sensitivity analysis was carried out on doses of up to 40 mg using the dose distribution from Tabas (2004).
- The studies included in the clinical effectiveness review did not stratify results by IBS subtype, so it was not possible to estimate the effectiveness for each of the subtypes separately. Therefore, it was assumed that these interventions are equally effective across all IBS subtypes.
Modelled response rates
Figures 18 to 23 below give the modelled response rates for six different strategies for prescribing tricyclics and SSRIs for the management of chronic pain in IBS.
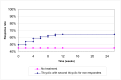
Figure 18
Response rates for a tricyclic up to 30 mg followed by second tricyclic up to 30 mg in non-responders compared to no treatment.
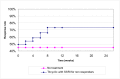
Figure 19
Response rates for a tricyclic up to 30 mg followed by an SSRI up to 20 mg in non-responders compared to no treatment.
Figure 20
Response rates for an SSRI up to 20 mg compared to no treatment.
Figure 21
Response rates for an SSRI up to 20 mg followed by a tricyclic up to 30 mg in non-responders compared to no treatment.
Figure 22
Response rates for an SSRI (up to 20 mg) followed by a second SSRI in non-responders compared to no treatment.
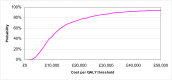
Figure 23
CEAC for up to 30 mg of tricyclic compared to no treatment.
- Tricyclic up to 30 mg
- Tricyclic up to 30 mg followed by second tricyclic up to 30 mg
- Tricyclic up to 30 mg followed by SSRI up to 20 mg
- SSRI up to 20 mg
- SSRI up to 20 mg followed by tricyclic
- SSRI up to 20 mg followed by second SSRI up to 20 mg
The RR of response for SSRIs versus placebo (or usual care) was much higher than the RR of response for tricyclics versus placebo (see Table 1) giving much larger increases in the number responding at each dose. However, as there was no head-to-head comparison of SSRIs and tricyclics, it was not possible to tell whether the apparent superiority of SSRIs is simply because they have been tested in a population that is more likely to respond to a pharmacological intervention. Indirect comparisons between SSRIs and tricyclics should be interpreted with caution due to the potential for bias.
Table 1
Intervention specific parameters – Tricyclics.
%20compared%20to%20no%20treatment.&p=BOOKS&id=51963_ch8f113.jpg "Click on image to zoom")
Figure 17Response rates for a tricyclic (up to 30 mg) compared to no treatment
Amitriptyline tablets were the lowest cost tricyclic at doses of 10 mg to 20 mg. Doxepin had a lower cost per 10 mg but the smallest tablet size is 50 mg, so it was not considered practical to assume that doxepin is commonly prescribed at a doses of 10 to 30 mg per day. It was assumed in the model that 10 mg amitriptyline tablets (non-proprietary) are prescribed as the first line tricyclic, with trimipramine (Surmontil®) prescribed second line. The highest cost tricyclic was amitriptyline solution (non-proprietary) at £5.03 per month for a dose of 10 mg per day, so this preparation was used in the high drug cost sensitivity analysis.
Of the two SSRIs considered, the lowest cost preparation is fluoxetine (non-proprietary) and it was assumed that this is prescribed first line, with the lowest cost paroxetine preparation (non-proprietary) prescribed second line. A sensitivity analysis was carried out assuming that the highest cost preparation is prescribed (Branded liquid preparations of fluoxetine and paroxetine at £14.40 and £9.62 per month respectively).
Table 2Intervention specific parameters – SSRIs
| Description | Value | Evidence | |
|---|---|---|---|
| RR of response for TCA vs placebo | 1.80 | Meta-analysis of RCT evidence for improvement in global symptoms | |
| Maximum number of switches considered | 1 to Tricyclic or second SSRI | Assumption based | |
| Drug costs | |||
| Intervention | Dose per day | Cost per month* (assuming lowest cost preparation) | |
| Fluoxetine | 10 mg to 20 mg | £0.75 to £1.50 | |
| Paroxetine | 10 mg to 20 mg | £3.05 to £6.10 | |
- *
British National Formulary (Joint Formulary Committee 2007)
RESULTS
Tricyclic up to 30 mg but no further treatment if no response
A strategy of using up to 30 mg of tricyclic is estimated to provide 0.46 additional QALYs (difference between QALYs gained for intervention and no treatment), at a cost of £4,459, compared to no treatment for a cohort of 100 patients over a 6 month period, provided treatment is not continued in those who do not respond to a dose of 30 mg. This strategy has a cost per QALY of £9,762, compared to no treatment, in the first 6 months after initiating treatment. Treatment can be continued in the next 6 months for a cost per QALY of £3,395 provided that treatment is reviewed every 6 months and discontinued in patients who no longer experience a therapeutic benefit, either due to lack of effectiveness or a change in their symptom profile.
The probabilistic sensitivity analysis estimates the uncertainty in the cost-effectiveness due to uncertainty surrounding the efficacy estimate, the utility gain, and the response rate in the no treatment arm. The results of this analysis are presented in Figure 24 as a CEAC, which shows the probability of the cost per QALY falling under various thresholds. The CEAC shows that a strategy of prescribing up to 30 mg of tricyclic has a 76% probability of being under £20K compared to no treatment over a 6 month timeframe.
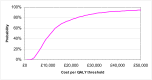
Figure 24
CEAC for up to 30mg a tricyclic with switch to a second tricyclic if no response compared to up to 30mg tricyclic without switch for non-responders.
Univariate sensitivity results for up to 30 mg of tricyclics
The basecase cost-effectiveness estimates assume that an equal number of patients respond to the 10 mg, 20 mg and 30 mg doses. However, if we assume that patients only respond after reaching the 30 mg dose, then the cost per QALY is higher at £11,296. If patients are allowed to increase their dose up to 50 mg in order to achieve a response and we assume that no patient responds to a lower dose then the cost per QALY is significantly higher at £17,937.
If the highest cost preparation is prescribed instead of the lowest cost preparation, then the cost per QALY is increased to £14,022. If the GP follow-up is twice as frequent as modelled in the pathway (i.e. every two weeks until an effective dose is established instead of every four weeks and two follow-up appointments in the next 12 weeks instead of one), then the cost per QALY is increased to £17,826. These sensitivity analyses suggest that using tricyclics at doses of up to 30 mg is likely to be cost-effective compared to no treatment, even if the costs of care are higher than estimated in the modelled care pathway.
The threshold analysis on the utility gain associated with an improvement in global symptoms shows that the cost per QALY would be over £20,000 if the utility gain is less than 0.035. For comparison, we used a utility gain of 0.071 in the basecase and a utility gain of 0.135 would be equivalent to a complete resolution of IBS symptoms.
Tricyclic up to 30 mg with switch to a second tricyclic if no response to first
If patients who do not respond to 30 mg of tricyclic are allowed to switch to a second tricyclic drug, and we assume that the second tricyclic is just as likely to be effective, then the cost rises to £5,858 per 100 patients over the first 6 months, but the total QALY gain is increased to 0.60. The cost per QALY of treating patients with up to two tricyclics to gain a response is £9,789 per QALY compared to no treatment, and £9,873 compared to stopping treatment after the first tricyclic. Even if the chance of response to the second tricyclic is half the chance of response to the first, the cost per QALY is £10,912 compared to no treatment and £18,324 compared to stopping after a failure on the first tricyclic.
A probabilistic sensitivity analysis was carried out on the cost-effectiveness of allowing non-responders to switch to a second tricyclic compared to no further tricyclic therapy for those who do not respond to the first tricyclic. The CEAC in Figure 25 shows that a strategy of prescribing up to 30mg of tricyclic with a switch to a second tricyclic for non-responders has an 87% likelihood of being under £20K compared to no further treatment for non-responders to 30mg of tricyclic.
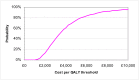
Figure 25
CEAC for up to 30mg of tricyclic with switch to an SSRI if no response compared to up to 30mg of tricyclic without switch for non-responders.
Tricyclic up to 30mg followed by an SSRI if no response to tricyclic
If patients who do not respond to 30mg of tricyclic are allowed to switch to an SSRI (up to 20mg), and we assume that the SSRI effectiveness is independent of the response to the tricyclic, then the cost rises to £5,648 per 100 patients over the first 6 months, but the total QALY gain is increased to 0.84. The cost per QALY of treating patients with a tricyclic at doses of up to 30mg followed by an SSRI of up to 20mg is £6,703 per QALY compared to no treatment, and £3,080 compared to stopping after the tricyclic. Even if the chance of response to the SSRI is half the chance of response seen in the SSRI trials, then the cost per QALY is £8,450 compared to no treatment and £5,342 compared to stopping after a failure on the tricyclic.
A probabilistic sensitivity analysis was carried out on the cost-effectiveness of allowing non-responders to switch to a SSRI compared to no further SSRI therapy for those who do not respond to the first tricyclic. The CEAC in Figure 26 shows that a strategy of prescribing up to 30mg of tricyclic with a switch to an SSRI for non-responders has a 95% likelihood of being under £10K compared to no further treatment for non-responders to 30mg of tricyclic.
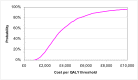
Figure 26
CEAC for SSRI up to 20mg (with no switching for non-responders) compared to no treatment.
SSRI up to 20mg but no further treatment if no response
A strategy of using up to 20mg of SSRI is estimated to provide 1.23 additional QALYs, at a cost of £3,708, compared to no treatment for a cohort of 100 patients over a 6 month period, provided treatment is not continued in those who do not respond to a dose of 20mg. This strategy has a cost per QALY of £3,020 compared to no treatment, in the first 6 months after initiating treatment. Treatment can be continued in the next 6 months for a cost per QALY of £1,483 provided that treatment is reviewed every 6 months and discontinued in patients who no longer experience a therapeutic benefit, either due to lack of effectiveness or a change in their symptom profile.
These estimates assume that an equal number of patients respond to the 10mg and 20mg doses. However, if we assume that patients only respond after reaching the 20mg dose, then the cost per QALY is higher at £3,209. If patients are allowed to increase their dose up to 40mg in order to achieve a response, then the cost per QALY is significantly higher at £3,790 when using the dose distribution from Tabas (2004). However, if no patient responds until a dose of 40mg is reached then the cost per QALY is higher still at £4,698.
If the highest cost preparation is prescribed instead of the lowest cost preparation, then the cost per QALY is increased to £9,799. If the GP follow up is twice as frequent as modelled in the pathway (i.e every two weeks until an effective dose is established instead of every four weeks and two follow-up appointments in the next 12 weeks instead of one), then the cost per QALY is increased to £5,667. These sensitivity analyses suggest that using SSRIs at doses of up to 20mg is likely to be cost-effective compared to no treatment, even if the costs of care are higher than estimated in the modelled care pathway.
The threshold analysis on the utility gain associated with an improvement in global symptoms shows that the cost per QALY would be over £20,000 if the utility gain is less than 0.011. For comparison, we have used a utility gain of 0.071 in the basecase and a utility gain of 0.135 would be equivalent to a complete resolution of IBS symptoms.
A probabilistic sensitivity analysis was carried out on the cost-effectiveness of treatment with an SSRI (up to 20mg) compared to no treatment. The CEAC in Figure 27 shows the probability of the cost per QALY falling under various thresholds. The CEAC shows that a strategy of prescribing up to 20mg of SSRI has a 95% likelihood of being under £10K compared to no treatment over a 6 month timeframe.
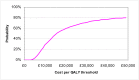
Figure 27
CEAC for up to 20mg of SSRI with switch to tricyclic if no response compared to up to 20mg SSRI without switch for non-responders.
SSRI up to 20mg with switching to a tricyclic (up to 30mg) if no response to SSRI
If patients who do not respond to 20mg of SSRI are allowed to switch to a tricyclic drug (up to 30mg), and we assume that the tricyclic effectiveness is independent of the response to the SSRI, then the cost rises to £4,526 per 100 patients over the first 6 months, but the total QALY gain is increased to 1.30. The cost per QALY of allowing patients to try an SSRI followed by a tricyclic is £3,477 per QALY compared to no treatment, and £11,073 compared to stopping after the SSRI. Even if the chance of response to the tricyclic in patients who haven’t responded to an SSRI is half the chance of response seen in the tricyclic trials, the cost per QALY is £3562 compared to no treatment and £21,574 compared to stopping after a failure on the first tricyclic. If the frequency of follow-up is twice that estimated in the basecase pathway, then the cost per QALY for treatment with an SSRI, followed by a TCA in non responders, is £6,523 compared to no treatment and £20,750 compared to stopping treatment after the SSRI.
A probabilistic sensitivity analysis was carried out on the cost-effectiveness of allowing non-responders to switch to a tricyclic compared to no further tricyclic therapy for those who do not respond to the SSRI. The CEAC in Figure 28 shows that a strategy of prescribing up to 20mg of SSRI with a switch to a tricyclic for non-responders has a 59.6% probability of being under £20K compared to no further treatment for non-responders to 20mg of SSRI. In the probabilistic analysis, there is a 12.8% probability that all of the patients respond to the first SSRI resulting in no further benefit to be gained by switching patients to a second SSRI. In drawing the CEAC we have assumed that the cost per QALY of allowing patients to switch would be above any reasonable threshold when no benefit can be achieved for that switch.
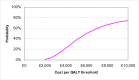
Figure 28
CEAC for up to 20mg SSRI with switch to a second SSRI if no response compared to up to 20mg SSRI without switch for non-responders.
SSRI up to 20mg with a second SSRI (up to 20mg) if no response to first SSRI
If patients who do not respond to 20mg of SSRI are allowed to switch to a second SSRI (up to 20mg), and we assume that the effectiveness of the second SSRI is independent of the response to the first SSRI, then the cost rises to £4,677 per 100 patients over the first 6 months, but the total QALY gain is increased to 1.43. The cost per QALY of allowing patients to try an SSRI followed by a tricyclic is £3,275 per QALY compared to no treatment, and £4,843 compared to stopping after the first SSRI. Even if the chance of response to the second SSRI in patients who haven’t responded to the first SSRI is half the chance of response seen in the SSRI trials, the cost per QALY is £3,361 compared to no treatment and £7,544 compared to stopping after a failure on the first SSRI.
A probabilistic sensitivity analysis was carried out on the cost-effectiveness of allowing non-responders to switch to a second SSRI compared to no further SSRI therapy for those who do not respond to the first SSRI. The CEAC in Figure 29 shows that a strategy of prescribing up to 20mg of SSRI with a switch to a second SSRI for non-responders has a 75% likelihood of being under £10K compared to no further treatment for non-responders to 20mg of SSRI. Again, there is a 12.8% probability that all of the patients respond to the first SSRI resulting in no further benefit to be gained by switching patients to a second SSRI. In drawing the CEAC we have assumed that the cost per QALY of allowing patients to switch would be above any reasonable threshold when there is no benefit can be achieved for that switch.

Indirect comparison of tricyclics and SSRIs
The results presented above (see Tables 3 and 4) for tricyclics compared to no treatment and SSRIs compared to no treatment, suggest that SSRIs are more cost-effective than tricyclics as they have a larger QALY gain and lower cost compared to usual care. However, as discussed earlier, this conclusion should be treated with caution as it is based on an indirect comparison and these have a high potential for bias.
Table 3
Results for up to 30 mg tricyclic (amitriptyline) with no switches compared to no treatment in a cohort of 100 patients with IBS (all subtypes).
Table 4
Results for up to 20mg SSRI (fluoxetine) with no switches compared to no treatment in a cohort of 100 patients with IBS (all subtypes).
EVIDENCE STATEMENTS
For this review, the evidence was assessed using the GRADE process and tables are shown in Appendix F. The following evidence statements are derived from the GRADE tables.
- There is a moderate amount of good quality evidence, mainly in patients with refractory IBS and with some depression, showing a significant global improvement in symptoms for both tricyclics and SSRIs when compared with placebo.
- There is limited evidence to show:
- A significant reduction in pain and bloating for tricyclics when compared with placebo.
- A borderline improvement in bowel habit for tricyclics when compared with placebo.
- There is a moderate amount of good quality evidence showing show no significant improvement in global symptoms for 50mg tricyclics (Trimipramine) compared with 30mg.
- The evidence is inconclusive as to whether there is an improvement in pain for SSRIs compared with placebo.
- There is a moderate amount of good quality evidence to show there is no significant reduction in bloating or improvement in bowel habit for SSRIs when compared with placebo.
- There is a moderate amount of weak evidence to show there is no significant improvement in quality of life for SSRIs when compared with usual care.
- There is a moderate amount of good quality evidence to show there are significantly more patients discontinuing treatment with SSRIs compared with usual care.
ADVERSE EFFECTS EVIDENCE STATEMENTS (BASED ON NICE CLINICAL GUIDELINE 23 ‘DEPRESSION’)
- 8.
Patients started on antidepressants who are not considered to be at increased risk of suicide should normally be seen after 4 weeks. Thereafter they should be seen on an appropriate and regular basis.
- 9.
In patients in primary care, there is insufficient evidence to determine whether there is a clinically significant difference between other antidepressants and amitriptyline (TCA) on reducing the likelihood of leaving treatment early either for any reason or due to side effects.
- 10.
There is good evidence in trials of eight weeks and longer, that there is no clinically significant difference between SSRIs and placebo on reducing the likelihood of leaving treatment early. This is not consistent when analysing the reasons for leaving treatment, which demonstrate a clinically significant difference favouring placebo over SSRIs in relation to leaving the treatment early due to side effects.
HEALTH ECONOMIC STATEMENT
Evidence from a decision analytic model showed that low dose tricyclics and SSRIs (trimipramine, amitriptyline, doxepin, paroxetine, fluoxetine) are cost-effective for long-term maintenance use in individuals with IBS. The cost-effectiveness estimate is based on a clinical pathway in which dose is increased gradually and response is assessed every four weeks until an effective dose is established or the maximum dose is reached. Trying a second tricyclic or SSRI in an individual who has not responded to a previous tricyclic or SSRI is also likely to be cost-effective when assuming that response to the second treatment is independent of a lack of response to the first. The cost-effectiveness analysis assumes that treatment is reviewed every 6 months to establish whether it is still relevant to the individual’s symptom profile.
EVIDENCE TO RECOMMENDATIONS
There is evidence from the clinical and cost effectiveness that tricyclics and SSRIs are effective in the symptom management of IBS. The GDG also took into consideration the reported adverse effects of tricyclics and SSRIs, but noted that the doses used for the management of IBS are similar to those used in the treatment of chronic pain and are thus much lower than starting doses used for the management of depression. The GDG was concerned that primary care clinicians consider the side effects when prescribing tricyclics and SSRIs. Although the reported side effects of tricyclics are more common (at higher doses) than SSRIs, the GDG reported that, in primary care, tricyclics are used in preference to SSRIs in low doses. Therefore the GDG advised that Tricyclics and SSRIs should be prescribed as second line treatment, starting with Tricyclics, and moving on to SSRIs only when the former had been shown to be ineffective.
RECOMMENDATION
Healthcare professionals should consider tricyclic antidepressants (TCAs)* as second-line treatment for people with IBS if laxatives, loperamide or antispasmodics have not helped. TCAs are primarily used for treatment of depression but are only recommended here for their analgesic effect. Treatment should be started at a low dose (5–10 mg equivalent of amitriptyline), which should be taken once at night and reviewed regularly. The dose may be increased, but does not usually need to exceed 30 mg.
RECOMMENDATION
Selective serotonin reuptake inhibitors (SSRIs) should be considered for people with IBS only if TCAs have been shown to be ineffective.
RECOMMENDATION
Healthcare professionals should take into account the possible side effects when prescribing TCAs or SSRIs***. After prescribing either of these drugs for the first time at low doses for the treatment of pain or discomfort in IBS, the person should be followed up after 4 weeks and then at 6–12 monthly intervals thereafter.
8.5. Adverse effects of pharmacological interventions
BACKGROUND
A wide variety of pharmacological interventions are available for the treatment of irritable bowel syndrome (IBS). As the various classes of agents have different pharmacological mechanisms of action, people with IBS may potentially be troubled by a wide range of adverse effects, depending on which treatment they are taking. While people with IBS are unlikely to experience significant harm from short-term or intermittent therapy, potential problems may arise if the drugs were taken over a longer-term period.
In making informed treatment decisions, health care professionals and people with IBS need to carefully weigh up evidence on the anticipated benefits against that of any relevant concerns about the safety and tolerability of IBS drug therapy. There are a few fundamental questions that can potentially be usefully answered from a review of adverse effects data:
- For people with IBS and health care professionals choosing to use a particular drug therapy for IBS, the review can inform them of potential adverse effects that could be anticipated from that therapy
- Availability of comparative data among different drugs can help in reaching a treatment decision based on which safety profile (or nature and frequency of adverse effects) is more acceptable.
While the overall frequencies of any adverse effects have been evaluated in the parallel efficacy reviews of IBS, there was limited detail given on what the specific adverse effects were, and whether the classes of drugs differ in their safety and tolerability profile.
8.5.1. Adverse effects: antispasmodics, antimotility agents and laxatives
SELECTION CRITERIA
The selection criteria described in the general methodology section were used, but some were specific to the evaluation of adverse effects and are reported in the following sections.
Types of studies
We did not apply any specific inclusion criteria based on study design; however, we preferred to exclude:
- Single case reports, as there was substantial scope for reporting and publication bias (towards the esoteric, interesting cases), and such cases may not have been representative of the general patient population
- Crossover studies, as it was impossible to discriminate between events that arise as a medium-long term complication of the first (previous) treatment, or as events resulting from the present therapy. This was particularly so as the protocol was primarily interested in evaluation of long-term effects from chronic administration of drugs for IBS. Moreover, adverse events are usually measured as dichotomous outcomes, (presence or absence of an adverse effect) and it was technically challenging to incorporate dichotomous measures from crossover studies into meta-analyses.
For the broad overall review, we accepted all studies evaluating the safety or tolerability of any drug therapy in populations of people with IBS.
We also looked at adverse effects for three specific classes of drugs for IBS; their selection criteria are listed below by each class of agent:
1. Antispasmodics
Interventions of interest:
- Dicyclomine bromide
- Hyoscamine (atropine)
- Hyoscine
- Alverine
- Mebeverine (including modified release)
- Peppermint oil.
Duration of intervention: minimum 4 weeks.
Outcomes
All outcomes reported within the categories of ‘adverse effects, side effects, adverse events, complications, safety, or tolerability’.
2. Antimotility Agents
Interventions of interest:
- Loperamide
- Co-phenotrope/Lomotil/Diphenoxylate.
Duration of intervention: minimum 4 weeks.
Outcomes
All outcomes reported within the categories of ‘adverse effects, side effects, adverse events, complications, safety, or tolerability’.
3. Laxatives
Types of participants
People with symptoms of IBS, including those with single symptom of IBS i.e. chronic constipation with no physical cause.
Excluded: studies of long-stay patients in hospital or nursing home setting, and palliative care patients (evidence too indirect as the groups are very different from people with IBS).
Interventions of interest:
- Polyethylene glycol (PEG) laxatives
- Lactulose.
Duration of intervention: minimum 4 weeks.
Outcomes
All outcomes reported within the categories of ‘adverse effects, side effects, adverse events, complications, safety, or tolerability’.
Quality of Adverse Effects Data
The techniques used in this review were generally based on advice within the Cochrane Handbook of Systematic Reviews regarding the assessment of adverse effects. This states, in particular, that the value of the data relies heavily on two major factors:
- How thorough were the methods used in monitoring adverse effects?
- How complete or detailed was the reporting?
In view of this, we concentrated on recording the following parameters:
- What methods (if any) did the trials stipulate for the specific assessment of AEs?
- Did the investigators pre-specify any possible adverse events that they were particularly looking out for?
- What categories of adverse effects were reported?
Identification of studies
We used a mixed strategy of checking articles that had already been retrieved for the efficacy reviews, and a new search of MEDLINE, EMBASE, CINAHL and the Cochrane Controlled Trials Register (search string: ‘drug-class’/ adverse-effects) with a total of 7206 hits. The search strategies are listed in Appendix B.
A total of 71 full text articles were screened; adverse effects data were extracted from 17 papers.
Study Design
The following types of studies were included in the adverse effects analysis:
- One non-randomised study which carried out an adverse effects survey in people with IBS
- Six randomised trials of antispasmodic agents
- One randomised trial of antimotility agents
- Seven randomised trials of laxatives (including one with a crossover design that should be considered with caution) and one observational long-term follow-up study.
Population
The underlying diagnosis was stated to be IBS for people in the observational study, as well as in the antispasmodic and antimotility agent trials.
However, the included laxative trials were predominantly in people with chronic or simple constipation, with no obvious physical cause. It is assumed that some of these people would have IBS.
Intervention and Comparisons
There was a very diverse range of interventions and associated comparator agents across the trials.
Assessment and Reporting of Adverse Effects
The results for the included trials are shown in Table 4 to Table 9.
Table 4
Details of RCTs of Antispasmodic agents.
Table 5
Details of RCTs of Antispasmodic agents.
Table 6
Details of RCTs of Antimotility agents.
Table 7
Details of RCTs of Antimotility agents.
Table 8
Details of RCTs on Laxatives.
Table 9
Details of RCTs of Antimotility agents.
Although some trials stated that they specifically enquired about adverse effects, none of them actually stated whether this was an open question e.g. ‘Are you having any problems with the treatment?’ of if the enquiry was targeted at particular symptoms e.g. ‘Have you had any diarrhoea?’
Moreover, some trials stated that people were asked to record problems in a diary, but the trial report gives no details about whether people had to record specific events (including when and how often) or if it was left to their discretion.
RESULTS
Overall Effects of Medication on people with IBS
Only one study looked at the overall impact of medication-related adverse effects on people with IBS (Lembo 2004). This was an online survey carried out in the United States, with people identified from a computer database. This survey collected data on medication use (questions on therapies used for symptoms of IBS) and self-reported assessment of adverse effects. Respondents were given a list of GI and non-GI adverse effects (including a free text entry box) and asked if they had experienced any side effects.
All respondents stated that they had been diagnosed with IBS by their physicians. Most of the participants were women (88%) with a median age of 45 years. The average number of medications they had tried was 3.3. Of the 668 respondents, 504 reported constipation as their primary symptom i.e. most people were of the IBS-C subtype.
The survey covered more than 10 drug classes, including laxatives, antispasmodics, antimotility agents, antidepressants etc., but the article reported mainly on laxatives and antispamodics. Overall 51% of people on antispasmodics complained of at least one adverse effect, in contrast to 59% of people on laxatives. Raw data from this survey was reported based on drug class, and the author did not specify what the individual agents and dosages were. The overall figures were:
Table 1
| Side Effect | Laxative | Antispasmodic | ||
|---|---|---|---|---|
| (n=171) | % | (n=189) | % | |
| Any | 100 | 58 | 96 | 51 |
| Drowsiness | 43 | 23 | ||
| Dizziness | 5 | 3 | 17 | 9 |
| Insomnia | 1 | 1 | 2 | 1 |
| Nausea | 32 | 19 | 4 | 2 |
| Abdominal cramps | 67 | 39 | 9 | 5 |
| Abdominal pain | 36 | 21 | 6 | 3 |
| Abdominal discomfort | 55 | 32 | 8 | 4 |
| Bloating | 36 | 21 | 6 | 3 |
| Dry mouth | 8 | 5 | 47 | 25 |
| Headache | 4 | 2 | 8 | 4 |
| Decreased sexual interest | 1 | 1 | 2 | 1 |
| Constipation | 7 | 4 | 9 | 5 |
| Diarrhea | 31 | 18 | 2 | 1 |
| None | 71 | 42 | 93 | 49 |
| SEVERITY | ||||
| Mild | 62 | 36 | 113 | 60 |
| Moderate | 94 | 55 | 55 | 29 |
| Severe | 14 | 8 | 21 | 11 |
The four most frequent adverse effects of antispasmodics are listed below. Figures for laxative users are given for purposes of comparison:
Table 2
| Antispasmodic (N= 189) | Laxative (N= 171) | Relative risk (95% CI) | |
|---|---|---|---|
| Dry mouth | 47 (25%) | 8 (5%) | 5.32 (2.59 – 10.94) |
| Drowsiness | 43 (23%) | 0 (0%) | Not estimable (Odds Ratio 8.65) |
| Dizziness | 17 (17%) | 6 (3%) | 2.56 (1.03–6.34) |
| Constipation | 9 (5%) | 7 (4%) | 1.16 (0.44–3.05) |
The six most frequent adverse effects of laxatives are listed below. Figures for antispasmodic users are given for purposes of comparison.
Table 3
| Laxative (N=171) | Antispasmodic (N=189) | Relative risk (95% CI) | |
|---|---|---|---|
| Abdominal cramp | 67 (39%) | 9 (5%) | 8.23 (4.24–16.0) |
| Abdominal discomfort | 56 (32%) | 8 (4%) | 7.74 (3.80–15.8) |
| Bloating | 36 (21%) | 6 (3%) | 6.63 (2.86–15.3) |
| Diarrhoea | 31 (18%) | 2 (1%) | 17.13 (4.16–70.5) |
| Abdominal pain | 36 (21%) | 6 (3%) | 6.63 (2.86–15.3) |
| Nausea | 33 (19%) | 4 (2%) | 9.12 (3.3 – 25.2) |
These findings indicated that laxatives wee significantly associated with GI adverse effects, while antispasmodics were associated with dry mouth, dizziness and drowsiness. The Relative Risk figures must be interpreted with caution as the treatment groups were not randomly allocated, and people with particular IBS symptoms may have been selectively channelled towards a specific class of treatment. However, it seems unlikely that channelling based on IBS subtype and existing gastrointestinal symptoms would have accounted for the marked difference seen with regards to side effects symptoms such as dry mouth, drowsiness and dizziness. Moreover, 504/668 (75%) of the respondents stated that constipation was their primary symptom. As the respondents had tried a mean of 3.9 IBS therapies, their adverse experiences with different classes of drugs could have been reflected within the survey responses.
Strengths of the study were that it surveyed people outside a trial setting, and they may potentially have been able to take the treatments over a longer time period. One important feature of the study was that it specifically enquired about certain adverse effects, covering both GI and non-GI problems.
The main weaknesses were that there was considerable potential for selection bias as the respondents may not have been typical of people with IBS. Recall bias was also a major problem if the treatments were taken a long time ago. We were also not certain of the diagnosis of IBS, as this was based on the respondents reporting that they had been diagnosed by a physician. As the survey was carried out in the US, larger or different dose regimens may have been used. Perhaps the most important limitation was that the individual agents were not listed, and lumping by drug class obscured any differences in the safety profiles of individual agents within a class.
RESULTS OF SPECIFIC CLASSES OF IBS THERAPIES
1. Antispasmodics
There were six included RCTs.
The interventions and comparators were extremely varied, as was the reporting of adverse effects outcomes. In view of this, no meta-analysis was performed and a descriptive summary is given in the appendix.
Mebeverine versus Dicyclomine
There was a trend towards a lower rate of adverse effects in the mebeverine group (RR 0.33, 95% CI 0.10, 1.04; p=0.06). GI symptoms were more frequently seen with dicyclomine.
Mebeverine standard preparation versus Mebeverine sustained release
No clear difference in safety or tolerability could be seen.
Mebeverine versus Trimebutine
Adverse effects were non-significantly more common in people taking mebeverine, with dry mouth being the most common complaint with mebeverine. Note: Trimebutine is not listed in the British National Formulary.
Alverine versus placebo
The authors reported 5 adverse events with alverine related to the nervous system but did not give any details.
2. Antimotility agents
We only identified two RCTs with adverse effects data. One was a crossover trial which should be treated with caution (Cann 1984).
No clear trend could be identified.
3. Laxatives
There were 7 included RCTs. Most of them did not specifically state that the participants had IBS, although it was very likely that some of the people with chronic constipation would fulfil the criteria for IBS.
The interventions and comparators were extremely varied, as was the reporting of adverse effects. A descriptive summary is given in the appendix.
Non-randomised study of TransiPEG
One long-term (6 month) observational study on 231 people taking TransiPEG provided limited information as there was no comparator group (Paille 1999). In this study, 21 people (9%) reported adverse effects with 14 (6%) stopping therapy due to adverse events. The most frequently reported problems were abdominal pain (8 people), flatulence (5 people), and diarrhoea (4 people).
Lactulose versus Isphagula husk
Two RCTs were evaluated, one of which was a crossover study (Quah 2006), and the findings must therefore be viewed cautiously. Abdominal pain occurred at a higher rate in the lactulose arms than the isphagula husk arms.
PEG versus lactulose (Ferguson and Attar 1999; Bouhnik 2004)
There were non-significant trends towards lactulose causing more abdominal pain and bloating, as compared to PEG. Flatus was also more common with lactulose (RR 1.72, 95% CI 0.99, 2.72).
PMF versus Placebo
No clear trend could be identified from the two small studies (Corazziari 1996; Corazziari 2000).
Different formulations of PEG
PEG 3350 and PEG 4000 showed similar rates of adverse effects. Low dose PEG in either form was associated with fewer adverse effects compared to high dose (Chaussade 2003).
The RCT data on lactulose was consistent with the findings of the non-randomised data with regards to increased risk of abdominal symptoms.
Limitations of the results
There were four major limitations that arose in this adverse effects review. The first problem was that the studies were primarily aimed at assessing and reporting on the efficacy of the drug treatments. Evaluation of safety took a back seat, and reporting of adverse effects data was often cursory or non-existent, even in instances where the methods sections had explicitly stated the intention of monitoring for adverse effects. Trial reports did not follow a structured format (e.g. by WHO system organ class) of reporting adverse effects, thus making it impossible to pool outcomes data.
The second major issue ws that none of the included studies appeared to anticipate, or pre-specify particular adverse effects of interest or concern. It was often possible to predict, based on pharmacological mode of action, the potential adverse effects of a drug therapy. Trial investigators could have designed specific aspects of the protocol to concentrate on detecting these adverse effects. While some of the trials did specify general measures for monitoring overall adverse effects, none of the reports stated whether they were checking for any specific problems. Given the wide-ranging nature of possible adverse effects, it may have been very difficult for trials to reliably pick up safety issues, unless there was some prior awareness of what the potential problems might have been.
The third major limitation was that many of the adverse outcomes of interest were very similar to the symptoms of the IBS itself. For instance, laxatives were associated with flatulence, cramps and abdominal pain – all of which are commonly seen in people with untreated IBS, and also form part of the efficacy assessment. This made it very difficult for the investigators to differentiate between the progress of the condition and the harmful effects of the drug. Moreover, some of these symptoms were listed within the efficacy section of the trial report, and it was not possible to determine whether a deterioration in these symptoms was due to lack of efficacy, the natural history of the condition, or the adverse effect of the drug.
The final limitation was the lack of specific non-randomised studies aimed at eliciting adverse effects of drug therapy in IBS. The only such study we looked at was not focused on particular drugs, and was only able to provide data on broad classes of IBS therapies, without naming specific drugs.
8.5.2. Adverse effects: tricyclics and selective serotonin re-uptake inhibitors
CRITERIA FOR CONSIDERING STUDIES FOR THIS REVIEW
Types of studies
Adverse effects data have been extracted from the randomised trials included in the clinical effectiveness review of antidepressants.
Search strategy for identification of studies
In discussion with colleagues at NICE, it was agreed that for adverse effects evidence, direct reference to Clinical Guideline 23 (Depression) was appropriate in order to supplement the RCT data. Contact with the National Collaborating Centre for Mental Health clinical effectiveness lead established the search strategy supporting their work, and evidence statements were lifted from the guideline in relation to the adverse effects for both TCAs and SSRIs.
PARAMETERS OF THE REVIEW
It is recognised that these drugs are typically used to treat large populations with psychiatric morbidities. The context for this review is to recognise the potential harmful effects of these drug types when prescribed at low dose for the treatment of IBS symptoms, namely pain and discomfort. In the last twenty years, tricyclics and SSRIs have been prescribed in the treatment of functional GI disorders such as IBS. The prevalence of anxiety and depressive disorders is high in people with severe and/or intractable IBS and may be present to some degree in all people with IBS. A recognised pharmokinetic effect from these drug types is an analgesic effect that is separate from inhibitor effects typically desired in the treatment of depression. There is growing evidence to suggest that visceral pain syndromes such as IBS may be effectively treated using these drugs, that appear to modulate the interactions between the central and enteric nervous systems.
The two drug classes (tricyclics, e.g. trimipramine, amitriptyline, doxepin, and SSRIs, e.g. paroxetine, fluoxetine) are among the medications that have been used as long-term maintenance therapy (i.e. for 3 months or more) for IBS.
Treatment (for example with amitriptyline, the lowest cost tricyclic) is generally initiated at 10mg with dose increases of 10mg no more frequently than every 2 weeks. Patients are encouraged to increase the dose until an effective dose is established or side-effects become problematic, up to a maximum dose of 30mg for tricyclics and 20mg for SSRIs. An exception to this is doxepin, where the smallest tablet size is 50mg. Patients who do not respond to maximum dose are switched to an alternative drug, with switches from first to second tricyclic considered and switches between tricyclics and SSRIs considered.
A. Tricyclics
1. Adverse effects
All TCAs cause, to varying degrees, anticholinergic side effects (dry mouth, blurred vision, constipation, urinary retention, sweating), sedation and postural hypotension. These side effects necessitate starting with a low dose and increasing slowly. Mianserin is a sedating tricyclic.
Evidence is available for the adverse effects occurring for doxepin (Boerner 1988) and mianserin (Tanum and Malt 1996). Tanum and Malt (1996) included the adverse effect of mild sedation, and it is known that mianserin is a sedating tricyclic. All the trials compared tricyclics with placebo control.
In Boerner (1988), a dose of 50mg doxepin was compared with placebo. In Tanum and Malt (1996), the dose of mianserin was up to 120mg; we note that in this study only 60% of the participants had IBS. These doses are higher than those generally recommended for the use of these drugs in IBS.

Figure 1
The Tanum and Malt (1996) study reported that significantly more patients were found to have adverse effects of mild sedation. In both studies the confidence intervals were wide.
One further study (Myren 1984) compared different doses of trimipramine (30 to 50mg). There were no significant differences between any of the trimipramine groups and placebo for palpitations, dizziness or dryness in the mouth. There was a significant increase in tiredness and morning drowsiness in the first 1 to 2 weeks for the antidepressant groups compared with placebo, but there were no significant differences at the end of treatment.
2. Number of patients withdrawing
Two studies recorded the numbers of withdrawals from the trials, for people receiving 120mg minaserin (Tanum and Malt 1996) and 10mg trimipramine (Tripathi 1983), compared with placebo. Meta-analyis showed a statistically significant effect in favour of placebo, but the confidence interval was wide.

Figure 2
B. SSRIs
1. Adverse effects
Evidence is available for the adverse effects occurring for fluoxetine, in comparison with placebo (Kuiken 2003). It was used at a dose of 20mg in this study. The confidence interval was fairly wide, but there was no significant difference between interventions.

Figure 3
2. Number of people withdrawing because of side effects
One study also investigated the number of participants withdrawing because of side effects for paroxetine compared with placebo (Tabas 2004); and Creed 2003 recorded the number who did not complete the treatment, for paroxetine compared with usual care. The confidence intervals were either wide or very wide, so conclusions could not be drawn about the non-significant results, but we noted there were significantly more people discontinuing treatment in the paroxetine group of the Creed (2003) study than in the usual care group. In Creed (2003), the dose of paroxetine was 20mg; and in Tabas (2004), the dose was up to 40mg.

Figure 4
C. Use of these drugs in the treatment of depression
Further evidence is available on the use of these medications (often at higher doses) for depression (NICE CG 23). For example, the effective dose of tricyclics for depression is usually taken to be 125mg. When used for depression, SSRIs are generally as effective as tricyclic antidepressants and are less likely to be discontinued because of side effects.
Tricyclics
Tricyclics are usually considered to have more side effects than other classes of drugs when used for depression, and people are less likely to stay in treatment on tricyclics than on other classes of drugs (RR= 0.71; 95% CI 0.65 to 0.78).
SSRIs
There is evidence suggesting that people are more likely to have side effects while on SSRIs than on placebo (RR=1.19; 95% CI 1.13 to 1.25), and are also more likely to withdraw from treatment due to side effects (RR=2.45; 95% CI 2.08 to 2.89). However, overall, a similar number of patients withdrew from treatment, so these withdrawals due to side effects of SSRIs may be offset by the number of people on placebo who withdrew for other reasons, for example due to lack of effectiveness of the treatment. Fewer people on SSRIs than on other antidepressants withdrew from treatment due to side effects (RR= 0.78; 95% CI, 0.71 to 0.85).
EVIDENCE STATEMENTS
Evidence statements for this review are mostly based on those in NICE Clinical Guideline 23 ‘Depression’ (statements 2 to 4).
- There is a moderate amount of good quality evidence to show there are significantly more patients discontinuing treatment with SSRIs compared with usual care.
- Patients started on antidepressants who are not considered to be at increased risk of suicide should normally be seen after 2 weeks. Thereafter they should be seen on an appropriate and regular basis.
- In people in primary care, there is insufficient evidence to determine whether there is a clinically significant difference between other antidepressants and amitriptyline (TCA) on reducing the likelihood of leaving treatment early either for any reason or due to side effects.
- There is good evidence in trials of eight weeks and longer that there is no clinically significant difference between SSRIs and placebo on reducing the likelihood of leaving treatment early. This is not consistent when analysing the reasons for leaving treatment, which demonstrate a clinically significant difference favouring placebo over SSRIs in relation to leaving the treatment early due to side effects.
Footnotes
- *
note that the trade name Dulco-lax is used for different drugs, but with different qualifiers.
- 2
In certain situations the daily dose of loperamide required may exceed 16 mg, which at the time of publication (February 2008) was an out of licence dose. Informed consent should be obtained and documented.
- *
At the time of publication (February 2008) TCAs did not have UK marketing authorisation for the indications described. Informed consent should be obtained and documented.
- **
At the time of publication (February 2008) SSRIs did not have UK marketing authorisation for the indication described. Informed consent should be obtained and documented.
- ***
At the time of publication (February 2008) SSRIs did not have UK marketing authorisation for the indication described. Informed consent should be obtained and documented.
- Pharmacological interventions - Irritable Bowel Syndrome in AdultsPharmacological interventions - Irritable Bowel Syndrome in Adults
- RecName: Full=Polyprenal reductase; AltName: Full=3-oxo-5-alpha-steroid 4-dehydr...RecName: Full=Polyprenal reductase; AltName: Full=3-oxo-5-alpha-steroid 4-dehydrogenase 3; AltName: Full=Steroid 5-alpha-reductase 2-like; AltName: Full=Steroid 5-alpha-reductase 3; Short=S5AR 3; Short=SR type 3gi|306526233|sp|Q9WUP4.2|SR5A3_MOUSProtein
- HSC34D092 normalized infant brain cDNA Homo sapiens cDNA clone c-34d09 3', mRNA ...HSC34D092 normalized infant brain cDNA Homo sapiens cDNA clone c-34d09 3', mRNA sequencegi|682175|gnl|dbEST|131890|emb|F096Nucleotide
Your browsing activity is empty.
Activity recording is turned off.
See more...