

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Tyrosyl-tRNA synthetase (TyrRS) comprises an N-terminal domain, which has the fold of the class I aminoacyl-tRNA synthetases, followed by idiosynchratic domains, which differ in eubacteria, archaebacteria and eukaryotes. The eubacterial TyrRSs have recruited an RNA binding domain which is found in a large family of proteins. The crystal structures of the TyrRSs from Bacillus stearothermophilus (Bst-TyrRS) and Thermus thermophilus (Tth-TyrRS) have been solved, free, or in complex with tyrosine, or with tyrosyl-αdenylate (Tyr-AMP). A quaternary complex between Tth-TyrRS, tRNATyr, tyrosinol and ATP has been solved at 2.8 Å resolution. The dimer of Bst-TyrRS is symmetrical in the crystals but asymmetrical in solution. It unfolds through a folded compact monomeric intermediate, by dissociation of the subunits (KD = 84 pM). A C-terminal domain is loosely linked to an intermediate α-helical domain through a fully flexible peptide. The tRNA binding site straddles the two subunits of TyrRS, which interacts with tRNATyr according to a class II mode. The conserved sequences of class I, HIGH and KMSKS, are involved in the catalysis of tyrosine activation. The HIGH sequence is not involved in the transfer of tyrosine from Tyr-AMP to tRNATyr, and the KMSKS sequence is involved in this transfer only through the initial binding of tRNATyr. Other residues (Thr40, Lys82 and Arg86 in Bst-TyrRS), are involved in both steps of the catalytic reaction, by interacting first with ATP then with residue Ade76 of tRNATyr. The identity elements of tRNATyr comprise nucleotidic base Ade73, the anticodon, and either base-pair Gua1:Cyt72 in eubacteria or Cyt1:Gua72 in archaebacteria and eukaryotes. The residues of TyrRS which interact with tRNATyr or recognize its identity elements have been identified by extensive mutagenesis and kinetic studies of Bst-TyrRS and from the structure of the Tth-TyrRS·tRNATyr complex. The two approaches are in excellent agreement. TyrRS catalyses the activation of tyrosine and its transfer to tRNATyr by stabilizing the transition states for these two reaction steps, through interactions with ATP, Ade76, and the identity elements of tRNATyr. The role of base pair 1:72 in the recognition of tRNATyr results in a species specificity and makes TyrRS a potential target for antibiotics. This specificity relies on a short segment (<41 residues) of TyrRS and can be swapped between species. The specific recognition between TyrRS and tRNATyr depends on the correct balance between the cellular concentrations of synthetases and tRNAs. Moreover, a residue (Glu152) of Bst-TyrRS is involved in the rejection of noncognate tRNAs but not in the interaction with tRNATyr, and thus is a purely negative determinant of specificity. Inhibitors of TyrRS have been discovered and characterized: tyrosinol, tyrosinyl-adenylate and tyrosyl-aryl dipeptides (e.g., Tyr-Tyr); however, they cross the bacterial envelope very inefficiently. Tyr-Gly dipeptides, derivatized with a sugar, have been isolated from microorganisms or synthesized, and shown to inhibit bacterial growth. The specificity of TyrRS towards the amino acid has been modified by screening or selecting mutants from random libraries which were targeted to residues of the tyrosine binding pocket. The species specificity of TyrRS towards tRNATyr and the absence of an editing mechanism towards the amino acid, have made it possible to create a 21st triplet of synthetase, tRNA and amino acid, and thus to extend the genetic code. TyrRS has additional functions in some organisms: it charges plant viral RNAs; acts as a kinase or a cytokine; plays a role in the splicing of the group I introns (as an RNA chaperone), in the maintenance of the mitochondrial genome, in yeast sporulation and in the quality control of tRNAs in the cell nucleus. TyrRS has been used in the synthesis of analgesic neuro-dipeptides.
Evolution of TyrRS
The tyrosyl-tRNA synthetases (TyrRS) of all the phylogenic kingdoms comprise an α/β domain, which has the mononucleotide binding fold of the class I aminoacyl-tRNA synthetases (aaRS) and an idiosynchratic insertion between the two halves of the fold (connective peptide CP1).1 In eubacteria, the α/β domain is followed by an α-helical domain, which is specific for TyrRS, and by a C-terminal domain, which belongs to a numerous family of RNA-binding domains.2 In the other two phylogenic kingdoms, the α/β domain is followed by a C-terminal (in archaebacteria) or intermediate (in eukaryotes) domain, which is common to TyrRS and to the tryptophanyl-tRNA synthetases (TrpRS) of all three kingdoms, and labelled C-W/Y. Finally, the Homo sapiens TyrRS (Hsa-TyrRS) comprises a C-terminal domain, homologous to the endothelial monocyte activating protein II (EMAP II), which is also found in eubacterial PheRS and MetRS.3 TyrRS and TrpRS appeared before the separation between eukaryotes and eubacteria, as the other aaRSs.4,5
Structure of TyrRS
The crystal structure of TyrRS from Bacillus stearothermophilus (Bst-TyrRS) has been solved at 2.3 Å resolution (fig. 1).The structures of complexes with tyrosine, tyrosinol, tyrosinyl-adenylate and tyrosyl-adenylate (Tyr-AMP) have also been reported. In the absence of tyrosine, ATP binds in the tyrosine binding pocket of TyrRS. In the presence of tyrosinol, which cannot be activated, ATP binds very weakly to the enzyme.6-9 Bst-TyrRS is a homodimer, possessing a two fold crystal symmetry. Each subunit of Bst-TyrRS comprises three sequential domains in the crystal structure. An α/β domain (residues 1-220), which contains the dimerization interface, and an α-helical domain (248-319) form a well ordered N-terminal domain (1-319). This domain is followed by an apparently disordered C-terminal domain (320-419). The properties of a Bst-TyrRS fragment, corresponding to the N-terminal domain, have shown that it is dimeric, it has the same crystal structure as the N-terminal domain in the full-length Bst-TyrRS, it forms Tyr-AMP with normal kinetics, but it does not bind tRNATyr (refs. 8, 10). The properties of a Bst-TyrRS fragment, corresponding to the C-terminal domain, were characterized in solution by biophysical techniques and compared to those of the full-length Bst-TyrRS and of its N-terminal fragment. These characterizations have shown that the C-terminal domain is not in a disordered state; on the contrary, that it is in a stable, condensed, defined state of folding; that it has similar structures in its isolated form and in the full-length Bst-TyrRS; and that its disorder in the crystals is due to the absence of tertiary interactions with the N-terminal domain.11,12

When the sequences of the known eubacterial TyrRSs are aligned and programs of secondary structure prediction applied, it becomes apparent that their C-terminal domains possess a precise and conserved secondary structure.13 The three-dimensional structure of the C-terminal fragment from B. stearothermophilus has been determined by NMR spectroscopy (fig.1).14,15 This C-terminal domain belongs to the super-family of the ribosomal protein S4 and its structure is new among the aminoacyl-tRNA synthetases.16-18
Recently, the structure of TyrRS from Thermus thermophilus (Tth-TyrRS) has been solved. According to the crystal form, none, one or two C-terminal domains are visible, in orientations that are different and determined by the crystal packing. The C-terminal domain of Tth-TyrRS is connected to the preceding α-helical domain by a linker peptide which is fully flexible in the absence of tRNA. In contrast, the C-terminal domain is stabilized in a fixed orientation in the structure of the complex between Tth-TyrRS and tRNATyr, and the linker peptide becomes more ordered.19
Stability of Eubacterial TyrRSs
The unfolding of the dimeric N-terminal fragment (residues 1-317) of Bst-TyrRS with urea was monitored by spectrofluorometry, circular dichroism and size exclusion chromatography. These experiments have shown the existence of an equilibrium between the native dimeric state of the N-terminal fragment, a folded monomeric intermediate and the unfolded polypeptide. From them, the stability of the association between the subunits (KD = 84 pM) and the stability of the monomeric intermediate were deduced. One third of the fragment stability comes from its dimeric nature, and the unfolding of the monomeric intermediate is about 3 times more cooperative than the dissociation of the subunits.20 As its monomer is inactive,21 TyrRS could have been formed by the association of two folded but inactive monomers during evolution.
To analyze how the stability of eubacterial TyrRSs has evolved, an ordered family of nine different hybrid proteins between TyrRS from the mesophilic bacterium Escherichia coli(Eco-TyrRS) and TyrRS from the thermophilic bacterium B. stearothermophilus (Bst-TyrRS) was constructed. The stability and activity of these hybrids were measured and their variations were analyzed when the position of the fusion point moved along the sequence. The results have shown that the two sequences can replace each other locally and still give stable and active TyrRS enzymes. The higher stability of Bst-TyrRS is due to cumulative changes of residues spread along the sequence, and compensatory changes of residues have occurred between the two sequences during evolution.22
The evolution of a particular tertiary interaction in Bst-TyrRS, at the crossing between helices H3 and H5 near the active site, was further studied. The residues involved, Ile52 and Leu105, are replaced with Leu and Val respectively in Eco-TyrRS. The corresponding mutations were constructed in Bst-TyrRS and their effects on stability measured. Mutation I52L destabilizes the association between the two subunits of Bst-TyrRS, even though residue Ile52 is located more than 20 Å away from the subunit interface. This observation has suggested that the same approach could allow one to study directly and quantitatively the effects of mutations, located around the active site, on the transfer of information through the subunit interface. Mutation L105V destabilizes the monomeric intermediate of unfolding. The two mutational pathways, going from the wild type Bst-TyrRS to the double mutant I52L-L105V, through one or the other of the two single mutants, are not equivalent. One of the pathways comprises two neutral steps for stability (I52L enabling L105V) whereas the other pathway comprises a destabilizing step (L105V) followed and compensated by a stabilizing step (I52L). Thus, the effects of mutations I52L and L105V on stability depend on the structural context. These results are compatible with the theory of the context effect in the evolution of proteins.23
Structure of the Complex between TyrRS and tRNATyr
Heterodimers between subunits of Bst-TyrRS carrying different mutations were constructed by in vitro methods and used to show that the binding site for one molecule of tRNATyr is shared between the two subunits of one TyrRS molecule (fig. 2).24,25 A structural model of the complex between Bst-TyrRS and tRNATyr was constructed and refined by successive cycles of predictions, mutagenesis to test these predictions, and molecular modeling. At the beginning, some basic residues of Bst-TyrRS were assumed to form salt bridges with phosphate groups of tRNATyr. The crystal structures of Bst-TyrRS and of tRNAPhe from yeast, obtained in their free states, were used for the modelings. Four residues of Bst-TyrRS involved in the binding of ATP, 13 residues belonging to the binding site of tRNATyr, and 1 residue (Glu152) involved only in the rejection of noncognate tRNAs were thus identified.25-29 The structural model of the complex between Bst-TyrRS and tRNATyr was published before any complex between a synthetase and a tRNA was solved by crystallography.25,26 In this model, the acceptor arm of tRNATyr interacts with the catalytic α/β domain of one subunit and its anticodon arm interacts with the C-terminal domain of the other subunit. TyrRS approaches tRNATyr by the side of its variable loop and the major groove of its acceptor stem, i. e. by the side opposite to that which was later shown to prevail among the class I synthetases.
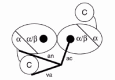
The crystal structure of the quaternary complex between Tth-TyrRS, its cognate tRNATyr, tyrosinol and ATP has recently been solved at 2.9 Å resolution (fig. 3 ).19 In this structure, the C-terminal domain is stabilized in a fixed orientation by binding in the elbow between the long variable arm and the anticodon stem of the tRNA. The tRNA binds across the two subunits of the dimeric enzyme and the mode of recognition resembles that of a class II synthetase, as predicted from the protein engineering studies on Bst-TyrRS. A comparison between the model structure of the Bst-TyrRS·tRNATyr complex and the crystal structure of the Tth-TyrRS·tRNATyr complex has shown that they are practically identical (at least for the interaction between the acceptor arm of tRNATyr and the N-terminal domain of TyrRS, the only one that could be modeled in the B. stearothermophilus system).30
Figure 3
Stereo view of the complex between T. thermophilus TyrRS, tRNATyr, tyrosinol and ATP, in the crystal structure at 2.9 Å resolution. Tyrosinol and ATP are shown in a space filling mode. Figure kindly provided by S. Cusack (see ref. 19).
Asymmetry of TyrRS in Solution
In the crystal structure of Bst-TyrRS, the two subunits are symmetrical through a two-fold rotational axis. The dimer binds two molecules of tyrosine and forms two molecules of Tyr-AMP.9 The dimer of Tth-TyrRS binds two molecules of tRNATyr in the crystals of the complex.19 In solution, Bst-TyrRS shows half-of-the-sites reactivity: the enzyme binds only one molecule of tyrosine or tRNATyr, and forms only one molecule of Tyr-AMP per molecule of dimer (for a review, see refs. 127, 32). Heterodimers between the full-length Bst-TyrRS and its N-terminal fragment were constructed in vitro (fig. 4 ). Such heterodimers comprise two sites for the formation of Tyr-AMP but only one site for the binding of tRNATyr. They can transfer tyrosine to tRNATyr only when Tyr-AMP is formed at the truncated subunit. It was observed that they charge tRNATyr at approximately half the rate of the parental Bst-TyrRS in the steady state. The following problem was therefore pinpointed. The stability of the enzyme bound Tyr-AMP combined with half-of-the-sites activity could lead to all of the enzyme accumulating as an inactive complex with Tyr-AMP bound at the full length subunit. Since the observed rate of charging does not decrease with time, there must be a mechanism for preventing random activation at one or the other active site, or selectively removing Tyr-AMP from the full-length subunit.25 The symmetry of Bst-TyrRS in solution was investigated by introducing different mutations in the full-length and in the truncated subunits of heterodimers. The results have shown that each dimer is active at only one site in solution, but that the site used is randomly distributed between the two subunits. No detectable interconversion is found between active and inactive sites over time. The wild type enzyme is thus inherently assymetrical in solution, even in the absence of substrate.32,33

Kinetics for the Formation of Tyr-AMP
The synthesis of Tyr-AMP from tyrosine and ATP, and then the transfer of tyrosine from Tyr-AMP to tRNATyr have been studied in great detail for Bst-TyrRS. For such studies, Bst-TyrRS mutants were constructed and their properties were compared to those of the wild-type synthetase in experiments of both steady-state and presteady state kinetics. The data on the synthesis of Tyr-AMP have been reviewed extensively.34-36 The strategy has mainly consisted in mutating residues of Bst-TyrRS that contact Tyr-AMP in the crystal structure of the complex.9 The lack of structure for the TyrRS·ATP and TyrRS·Tyr-AMP·PPi complexes, and the lack of well defined bonds between Bst-TyrRS and the adenine ring of Tyr-AMP have first hampered the elucidation of the mechanism for Tyr-AMP synthesis.37 However, two types of analyses made further progress possible.
1) Chemical studies have shown that the formation of Tyr-AMP from tyrosine and ATP results in the inversion of the α-phosphorus, that no covalent enzyme-bound intermediate occurs and that the mechanism involves an “in-line” displacement, with the tyrosyl carboxylate acting as a nucleophile and the pyrophosphate being the leaving group. Such a reaction scheme implies a penta-coordinated transition state.38 By using this chemical information, Leatherbarrow and coworkers constructed a model of the penta-coordinated intermediate between ATP and tyrosine.39 This model suggested that Thr40 and His45 are involved in the binding of ATP. Residues His45 belongs to a sequence motif, HIGH, which is conserved among the class I aaRSs.40,41
2) A systematic mutagenesis of the positively charged residues of Bst-TyrRS has shown that the changes of four residues in its N-terminal domain, Lys82, Arg86, Lys230 and Lys233, strongly affect the rates of Tyr-AMP synthesis and pyrophosphate exchange.25 This result was unexpected because these four residues are far from Tyr-AMP in the crystal structure of Bst-TyrRS. Two of these residues, Lys230 and Lys233, belong to a pentapeptide which is conserved among the sequences of the class I aaRSs.41,42 The mutagenesis results have thus established the role for this motif, KMSKS, in the reaction of amino-acid activation.
Detailed kinetic studies have subsequently shown the role of residues Thr40, His45, Lys82, Arg86, Lys230, Lys233 and Thr234 in the binding of the β- and γ-phosphates of ATP, and in the binding of pyrophosphate at different steps in the formation of Tyr-AMP (see chapter by E. First in this book).36
Kinetics for the Transfer of Tyrosine
Detailed kinetic studies have been performed on the transfer of tyrosine from the Tyr-AMP intermediate to the 3'-end of tRNATyr by Bst-TyrRS. Four species of tRNATyr were assayed as substrates for Bst-TyrRS and found to be equivalent in the in vitro charging reaction: native E. coli tRNATyr, native B. stearothermophilus tRNATyr, B. stearothermophilus tRNATyr expressed in E. coli, and B. stearothermophilus tRNATyr transcribed in vitro. Therefore, the modifications of tRNATyr are not essential for its recognition by Bst-TyrRS in vitro.43
The free energies of two molecular species, coming after the intermediate TyrRS·Tyr-AMP in the reaction pathway of Bst-TyrRS, have been determined by experiments of presteady state kinetics: those of the initial complex TyrRS·Tyr-AMP·tRNATyr and of the transition state complex TyrRS·[Tyr-tRNATyr-AMP]. These determinations have shown that the activation energies of the two chemical steps in the reaction pathway, (a) the activation of tyrosine and (b) its transfer to tRNA, are of equal intensity: 15.4 kcal·mol-1. Therefore, the optimization of the global catalysis by TyrRS during evolution has probably involved a selective pressure on both steps.43 To test this assumption, the rate constants for the transfer of tyrosine from Tyr-AMP to tRNATyr by Bst-TyrRS mutants were measured. Mutations of Thr51 were chosen because this residue contributes to the rate constant for the activation of tyrosine, and to the binding of Tyr-AMP. The rate constant for the transfer of tyrosine decreased when the stability of the TyrRS·Tyr-AMP complex increased, due to mutations of Thr51. The stabilization of the TyrRS·Tyr-AMP complex by a mutation of Thr51 therefore accelerated the activation step but slowed down the transfer step of the aminoacylation reaction, consistent with the tested assumption.44
The role of the conserved HIGH sequence in the catalysis of the transfer step was analyzed using the H45A and H48A mutants of Bst-TyrRS. This analysis has shown that the HIGH sequence is involved in the activation step but not in the transfer step, i. e. the kinetic parameters for the transfer of tyrosine to tRNATyr are not affected by the mutations. In contrast, residue Thr40 which is present in a subset of the class I aaRSs, and Lys82 and Arg86 which are present only in TyrRSs, stabilize the transition states for both reaction steps.45 The role of the conserved KMSKS sequence in the catalysis of the transfer step was analyzed similarly. Although Lys230 and Lys233 of Bst-TyrRS are involved in the initial binding of tRNATyr and act synergistically, none of the residues in the KMSKS sequence affect the rate constant of the transfer step.46 From an evolutionary viewpoint, the results on the HIGH and KMSKS motifs of Bst-TyrRS have suggested that the stabilization of the transition state for the first step of the aminoacylation reaction has preceded that for the second step. They are consistent with the proposal that the capacity of the aaRSs to catalyse the activation of the amino acid has preceded their capacity to attach the amino acids to the 3'-OH end of tRNAs.45
During the construction of the structural model for the complex between Bst-TyrRS and tRNATyr, rotations around the bonds which bracket phosphates-74, -75 and -76 were allowed, to bring the 2'-OH and 3'-OH of adenosine 76 (Ade76) in position to attack the carbonyl carbon of Tyr-AMP. In this model, Ade76 is located in the vicinity of residues Thr40, Lys82 and Arg86.26 The kinetic data on mutations T40A, K82A and R86A are consistent with such a model and suggest that residues Thr40 and Lys82 each form one hydrogen bond (H-bond) with Ade76, whereas Arg86 forms two H-bonds with it (Table 1). Mutations T40A, K82A and R86A have a small effect on the initial binding of tRNATyr (0.5 to 1.0 kcal·mol-1) and a much larger effect on the stability of the transition state (1.7-3.5 kcal·mol-1). These data suggest that a conformational change could take place during the formation of the transition state for the transfer reaction, and that the main role of Thr40, Lys82 and Arg86 could be to position Ade76 of tRNATyr correctly for an attack on Tyr-AMP.45
Table 1
Interaction energies of side chains of Bst-TyrRs with tRNAtyr.
Residues Thr40, Lys82 and Arg86 have similar roles in the first and second steps of the aminoacylation reaction. They contribute little binding energy to the formation of the initial state complex TyrRS.Tyr.ATP, and a more important energy to the stabilisation of the transition state complex TyrRS·[Tyr·ATP]. The three residues contribute little energy to the interaction with Ade76 of tRNATyr during the formation of the initial state complex TyrRS·Tyr-AMP·tRNATyr and an important energy to the stabilization of the transition state complex TyrRS·[Tyr-tRNATyr-AMP]. The three residues interact with the β- and γ-phophates of ATP in the first step, and probably with the ribose portion of Ade76 in the second step. Globally, the data are compatible with a catalytic mechanism in which TyrRS catalyses the activation of tyrosine and its transfer to tRNATyr by bringing together the substrates in the right orientation for the reaction to occur, and by utilizing the binding energy provided by noncovalent interactions to preferentially stabilize the transition states.34,45
Several catalytic mechanisms have been evaluated for the transfer reaction. Residue Gln195 forms a H-bond with the carbonyl oxygen of Tyr-AMP and Gln173 forms a H-bond with the protonated ammonium group of Tyr-AMP in the crystal structure of the Bst-TyrRS·Tyr-AMP complex. Thr40, Lys82 and Arg86 probably interact with Ade76 during the second step of the reaction. Kinetic experiments in the presteady state have shown that mutation Q195A has no effect on the binding or rate constants for the transfer reaction. Mutations of the four other residues, Thr40, Lys82, Arg86 and Gln173, into Ala affect little or not the formation of the initial complex TyrRS·Tyr-AMP·tRNATyr and strongly destabilize the transition state complex TyrRS·[Tyr-tRNATyr-AMP]. Thus, the stabilization of the transition state by TyrRS involves side chains (Thr40, Lys82 and Arg86) which are important for the formation of the new bond between tyrosine and Ade76, and a side chain (Gln173) which is probably important for the cleavage of the acyl-phosphate bond. On the basis of these data, First and coworkers have suggested that the transfer reaction occurs through a concerted mechanism in which the cleavable acyl-phosphate bond is elongated and strained, and the bond between the carbonyl carbon and the 3'-OH or 2'-OH of ribose 76 is partially formed.47 The role of various residues in the transfer reaction is summarized in (Table 1.)
Identity Elements of tRNATyr
Eubacteria. The identity elements of tRNATyr have not been analyzed systematically in eubacterial systems, in particular by transplantation into a noncognate tRNA. The in vitro properties of chemically modified E. coli tRNATyr molecules (Eco-tRNATyr) and the in vivo properties of mutant derivatives of the amber suppressor Eco-tRNATyr(CUA) have provided a first delineation of the nucleotide residues through which Eco-tRNATyr is recognized by Eco-TyrRS.31,48,49 A long variable stem and loop may not be absolutely required for recognition by Eco-TyrRS since a mutant Eco-tRNACys, carrying the native Gua1:Cyt72 base pair, the amber anticodon CUA, and mutation U73A, inserts tyrosine into dihydrofolate reductase in vivo.50 The equivalence of native and in vitro transcribed Eco-tRNATyr molecules for in vitro tyrosylation by Eco-TyrRS has been established. Steady-state kinetics for the charging of Eco-tRNATyr variants by Eco-TyrRS in vitro have shown that the recognition elements of Eco-tRNATyr include Gua34, Uri35, the orientation of the long variable loop (dependent on the unpaired residues Uri46 and Uri47), and Ade73. Base pair Gua1:Cyt72 is recognized with a weak preference (Table 2).51-53
Table 2
Effects of mutations in the identity elements of Eco-tRNATyr on charging by Eco-TyrRSa.
Archaebacteria. Mutagenesis and transplantation experiments have shown that the identity elements of Methanococcus janaschii tRNATyr (Mja-tRNATyr) comprise Ade73 as the most important element, base pair Cyt1:Gua72, and the anticodon with mainly Gua74. This set of identity elements is complete.54,55
Saccharomyces cerevisiae. The native and the in-vitro transcribed cytoplasmic S. cerevisiae tRNATyr (Sce(cyt)-tRNATyr) have similar kinetics of tyrosylation by Sce(cyt)-TyrRS in vitro. Mutagenesis and transplantation experiments have shown that the identity elements of Sce(cyt)-tRNATyr comprise Ade73, the base pair Cyt1:Gua72, and the anticodon with mainly Gua34. This set is complete.56 The change of pseudo-Uri35 into several modified pyrimidines has shown that the two N-H groups of this residue are requested for an optimal interaction with Sce(cyt)-TyrRS.57
Recognition of tRNATyr and Its Identity Elements by TyrRS
The main regions of Bst-TyrRS and Tth-TyrRS which contact their cognate tRNATyr are summarized in (Table 3). For the B. stearothermophilus system, these contacts have been deduced from mutagenesis experiments and structure modelings.25-27,29 For the T. thermophilus system, they have been deduced from the crystal structure of the complex.19 There is an excellent agreement between the two complementary approaches. The regions of contact include the four clusters of basic residues which have been found by mutagenesis studies.25,26,58 The contributions of the contact residues to the stabilities of the initial and transition state complexes have been evaluated from the kinetic parameters for the tyrosylation of tRNATyr by Bst-TyrRS mutants, in steady state experiments (Table 4).
Table 3
Comparison of the regions in contact with tRNATyr in the model structure for Bst-TyrRs and in the crystal structure for Tth-TyrRS,,,.
Table 4
Apparent destabilization of the initial complex(ΔΔGs) and of the transition state complex (ΔΔGT) for the charging of Eco-tRNATyr by Bst-TyrRSa.
The interactions and the mechanism by which TyrRS recognizes the identity elements of tRNATyr have been deduced from the above data and the tyrosylation of tRNATyr variants by TyrRS in the E. coli system, in steady state kinetics experiments (Table 5). In particular, the recognition of the discriminator base Ade73 involves residues of the connective peptide CP1, and the recognition of the anticodon involves a residue located in the peptide linking the α-helical and C-terminal domains. Comparison of the kinetic parameters KM and kcat/KM for the charging of Eco-tRNATyr variants has suggested that the identity elements form stronger interactions with Eco-TyrRS in the complex of the transition state TyrRS·[Tyr-tRNATyr-AMP] than in the complex of the initial state TyrRS·Tyr-AMP·tRNATyr. Their recognition by TyrRS would thus stabilize the transition state for the transfer reaction and decrease the corresponding activation energy (Table 2). Conversely, the kinetic parameters for the charging of tRNATyr by Bst-TyrRS variants leads to the same conclusions. For example, mutation K151N of Bst-TyrRS does not affect KM(tRNATyr) but decreases strongly kcat/KM for the charging reaction. Similarly, mutation W196A affects more kcat/KM than KM(tRNATyr).25,26 The mutations of the discriminator nucleotide Ade73 of Eco-tRNATyr affect more kcat/KM than KM(tRNATyr) for the aminoacylation by Eco-TyrRS.52 Thus, Ade73 appears to be fully recognized by TyrRS only in the transition state for the charging of tRNATyr (ref. 59). These conclusions were deduced from experiments of steady state kinetics for a reaction which comprises two steps.60 However, the two steps have the same activation energy (at least for Bst-TyrRS) and the presence of tRNATyr does not affect the activation of tyrosine.32
Table 5
Recognition of the identity nucleotides of tRNATyr by Bst-TyrRS and Tth-TyrRS,,.
The observation that mutations of the tRNATyr anticodon, or mutations of Bst-TyrRS residues which interact with the anticodon arm affect the kcat parameter for tyrosylation, suggests that there is a transmission of information between the distal regions of either tRNATyr or TyrRS, and either the acceptor end of the tRNA or the active site of the synthetase. This transmission could take place through either the tRNA or the synthetase, with or without a conformational change.
Residue Gua35 is a minor identity element of Mja-tRNATyr (ref. 55). Residues Asp286 and Lys288 of Mja-TyrRS belong to a motif of the C-terminal domain, PXDLK, which is conserved in the eukaryotic and archaebacterial TyrRSs. Mutations D286A and K288A do not affect the tyrosylation of a minihelixTyr but they do affect the charging of a full-length tRNATyr. Moreover, mutations K288A in Mja-TyrRS and U35G in Mja-tRNATyr have nonadditive effects on the charging reaction.54 These data suggest an interaction between Lys288 and the anticodon, contrary to the authors' conclusion.
Lee and RajBhandary were the first to deduce that the specific recognition of base pair Cyt1:Gua72 relative to Gua1:Cyt72 in Sce(cyt)-tRNATyr, implies that this base pair is recognized by Sce(cyt)-TyrRS in the major groove of the helical acceptor stem.61
Species Specificity
The presence of base pair Gua1:Cyt72 in eubacteria and Cyt1-Gua72 in archaebacteria and eukaryotes as identity elements, results in a species specificity. It was observed 30 years ago that the S. cerevisiae mitochondrial tRNATyr (Sce(mit)-tRNATyr) can be tyrosylated by Eco-TyrRS in vitro and in vivo.62,63 In contrast, an amber suppressor tRNA(CUA), which carries base Ade73 and base pair Gua1:Cyt72 as the eubacterial tRNATyr, cannot be charged by Sce(cyt)-TyrRS in vivo.64 However, it can be charged both in vivo and in vitro if it carries Cyt1-Gua72.61 Similar conclusions were obtained by comparing the charging of microhelicesTyr, derived from S. cerevisiae or E. coli tRNATyr, by Eco-TyrRS and the TyrRS from the lower eukaryote Pneumocystis carinii. These microhelicesTyr carried either Gua1:Cyt72 or Cyt1-Gua72 in addition to Ade73. In particular, the charging of the E. coli minihelixTyr by Eco-TyrRS is weakly affected by the change of base pair Gua1:Cyt72 into Cyt1-Gua72.53 As regards higher eukaryotes, a recombinant Hsa-TyRS charges Hsa-tRNATyr but not Bst-tRNATyr, whereas the reciprocal is true for Bst-TyrRS.65 The systematic characterization of the identity elements for tRNATyr in the M. janashii and S. cerevisiae systems has firmly established the molecular basis of this species specificity,55,56 which might be an important property for the use of TyrRS as a target for new antibiotics.
Mycobacterium tuberculosis TyrRS can charge Eco-tRNATyr in vivo and in vitro, but does not function in the S. cerevisiae cytoplasm. The comparison of its sequence with those of other eubacterial and eukaryotic TyrRSs revealed that the species specificity of TyrRS towards tRNATyr is encoded non only in the identity elements of the tRNA (Cyt1-Gua72 versus Gua1-Cyt72) but also in TyrRS.58 For example, the replacement of a peptide of 41 residues in Eco-TyrRS (residues 129-172, corresponding to 126-166 in Bst-TyrRS) by the homologous peptide from Hsa-TyrRS (125-162) enables the charging of Sce(cyt)-tRNATyr by Eco-TyrRS. The reciprocal result, charging of Eco-tRNATyr by an engineered Hsa-TyrRS, is also true.66 In Bst-TyrRS, this peptide comprises residues Asn146, Ala150, Lys151 and Glu152, which are important either for the specific recognition of tRNATyr or for the rejection of noncognate tRNAs.26,28
Sequence comparisons have shown that three residues which stabilize the transition state for the formation of Tyr-AMP by Bst-TyrRS (Cys35, His48 and Lys233) are not present in Hsa-TyrRS. Moreover, Hsa-TyrRS needs potassium ions for activity, contrary to Bst-TyrRS. Presteady state kinetics experiments have shown that the two enzymes have identical activation energies for the synthesis of Tyr-AMP, despite the differences between their active sites and their requirements for the K+ ion. The differences between the two enzymes could be exploited for the rational design of antibiotics (J. Austin & E. A. First, submitted).
Discrimination between tRNAs
Correct aminoacylation depends not only on identity elements in tRNAs and their recognition by their cognate aaRSs, but also on competitions between different aaRSs for a given tRNA, or different tRNAs for a given aaRS.67 Several studies with the tyrosine system have substantiated this concept of competition.
When Eco-GlnRS is overproduced in vivo, it incorrectly acylates the amber suppressor tRNATyr(CUA) with glutamine. This mis-aminoacylation is abolished if the intracellular concentration of the cognate Eco-tRNAGln is increased.68 It is also abolished if Eco-TyrRS is overproduced in vivo. This last effect can be reproduced in vitro: TyrRS competes with GlnRS for tRNATyr(CUA) charging with glutamine. These experiments have shown that the competition between the two aaRSs occurs at the aminoacylation step and suggested that it depends on their relative affinities for the tRNA.49 Similarly, although the mutant amber suppressor Eco-tRNATyr(G3:U70, CUA) is quantitatively aminoacylated with both alanine and tyrosine in vitro, competition between Eco-AlaRS and Eco-TyrRS prevents aminoacylation with alanine in vivo. As the concentration of the AlaRS increases, the identity of tRNATyr(G3:U70, CUA) is switched from a tyrosine to an alanine tRNA.51
The overproduction of Eco-TyrRS or Bst-TyrRS is toxic for E. coli and results in the destabilization of cellular proteins. The toxicity increases with the growth temperature. The causes of this phenomenon were analyzed by varying the cellular concentrations of TyrRS and its activity of tRNATyr charging, through genetic means. These experiments have shown that the toxicity of the overproduced TyrRS results from its interaction with tRNAs and probably from the ensuing mis-incorporation of amino acids into essential proteins. They have also shown that the balance between the cellular concentrations of the aaRSs and tRNAs is essential for the cell viability, by contributing to the precision of the translation of the genetic code.31 Similarly, Sce(cyt)-TyrRS is not toxic for E. coli when it is produced from a vector of low copy number and at low temperatures (22 °C to 30 °C). However, it becomes toxic at high cellular concentrations or growth temperatures. The toxicity is due to the charging of Eco-tRNAPro with tyrosine.69
During the genetic translation, each aaRS specifically aminoacylates its cognate tRNAs and rejects the 19 other species of tRNAs. A decrease in the specificity of this reaction can lead to mis-incorporations of amino acids into proteins and be deleterious to the cell. Residue Glu152 of Bst-TyrRS is close to phosphate groups 73 and 74 of tRNATyr in the structural model of their complex. Eleven changes of Glu152 were created by mutagenesis to determine whether this residue contributes to the recognition of tRNATyr and to the discrimination between tRNAs by Bst-TyrRS. The mis-aminoacylations of tRNAPhe and tRNAVal with tyrosine in vitro (on a scale going from 1 to 30) and the toxicity of Bst-TyrRS in vivo (on a scale going from 1 to 107) increased in a correlated way when the nature of the side chain in position 152 varied from negatively charged to neutral then to positively charged. The aminoacylation of tRNATyr was not affected by the mutations. The toxicity of the mutations was abolished by a second mutation in TyrRS, which prevents the binding of tRNATyr. These results have shown that the role of Glu152 in the discrimination between tRNAs is purely negative and that it acts by electrostatic repulsion of the noncognate tRNAs.27,28 Such a role is supported by the structure of the Tth-TyrRS·tRNATyr complex.19
Inhibitors
As TyrRS is an essential cellular protein, inhibitors could be used as antibiotics. Stabilized mimics of Tyr-AMP, such as tyrosinyl adenylate, are potent inhibitors of TyrRS but their polarity prevents their transport across the bacterial cell wall.6,70 Several triazine dyes inactivate Bst-TyrRS irreversibly. They are excluded from the tyrosine binding site and occupy the ATP-binding site. These dyes are not specific for TyrRS and also inactivate TrpRS and MetRS.71,72 Tyrosyl aryl dipeptides, which inhibit the aminoacylation activity of Staphylococcus aureus TyrRS (Sau-TyrRS) (IC50 = 0.5 μM) have been identified. A crystal structure of Sau-TyrRS complexed with one of the inhibitors, the dipeptide Tyr-Tyr, shows occupancy of the tyrosine binding pocket and interactions of the inhibitor with key catalytic residues.73
A potent inhibitor, specific for bacterial TyrRSs and designated SB-219383, has been isolated from a Micromonospora species. SB-219383 shows competitive inhibitory activity against Sau-TyrRS (Ki = IC50 = 0.6 nM for Sau-TyrRS; IC50 = 22 μM for mammalian TyrRS) and weak anti-bacterial activity against some Streptococcal strains in vitro (MIC = 32 μg/ml).74 SB-219383 can be described as a Tyr-Gly dipeptide, in which the Cα-position of the Gly moiety is derivatized with a bicyclic sugar.75 Several derivatives of SB-219383, which retain high inhibitory activities, have been synthesized. SB-239629 (IC50 = 3 nM) is a monocyclic derivative of SB-219383, obtained by cleaving its bicyclic sugar; SB-243545 (IC50 = 0.3 nM) is a butyl ester derivative of SB-239629; in SB-284485 (IC50 = 4 nM), the bicyclic sugar of SB219383 is replaced with fucose. The crystal structures of complexes between Sau-TyrRS and each of these four inhibitors have been solved, down to 2.2 Å resolution for some of them. The bicyclic sugar of SB-219383 and its monocyclic derivative in SB-239629 globally occupy the binding site of TyrRS for ribose. The butyl group of SB-243545 has revealed the existence of a new binding pocket in TyrRS, which involves displacements of the HIGH and KMSKS class I motifs. The fucose moiety of SB-284485 forms more H-bonds with TyrRS than the ribose moiety of Tyr-AMP. These structures have revealed the existence of five different binding sites in TyrRS (for tyrosine, a-phosphate, ribose, adenine, butyl and pyrophosphate) which could be further explored for the design of inhibitors.76 Other pyranosyl and carbocyclic analogs of SB-219383 have been synthesized to reduce its overall polarity and thus improve its penetration through the bacterial cell wall. One of the compounds shows as high an inhibitory activity against Sau-TyrRS as SB-219383 and an improved antibacterial activity against Moraxella catarrhalis and Streptococcus pyogenes (MIC = 8 μg/ml).77,78
From random libraries displayed at the surface of phage M13, peptides that bind to Haemophilus influenzae TyrRS (Hin-TyrRS) have been isolated. Most of these peptides are specific inhibitors of the Hin-TyrRS activity and appear to preferentially bind to the TyrRS active site. One of the corresponding synthetic peptides showed a competitive inhibition towards tyrosine (Ki = 80 nM) and a mixed inhibition towards ATP (Ki = 60 nM). Another showed noncompetitive inhibitions towards both tyrosine and ATP (Ki = 300-500 nM). These two peptides were used in a binding assay to detect small inhibitory molecules, in the μM to nM range, that bind to the same sites.79
Charging of Noncognate or Nonnatural Amino Acids
The mechanism by which Bst-TyrRS specifically recognizes tyrosine was studied by mutagenesis of residues close to the active site, according to the crystal structures. The carboxylate of Asp176 makes a hydrogen bond with the hydroxyl group of tyrosine.9 Mutation of Asp176 results in an inactive enzyme. Asn123 and Trp126 do not interact directly with tyrosine but appear to make H-bonds with Asp176. Mutations N123A and N123D strongly affect the kinetics of tyrosine activation, and in particular kcat and KM(Tyr). In contrast, W126F and W126L do not affect these kinetics with respect to ATP, and modestly increase KM(Tyr). The specificity for Tyr against Phe, determined from the ratio kcat/KM in the pyrophosphate exchange reaction (1.2·105 for the wild type TyrRS), decreases 4 fold for N123A but increases 2 fold for W126L and 7 fold for W126F. Thus, the wild type enzyme can be improved for discrimination between Tyr and Phe.80
An advantage of TyrRS, when looking for the charging of nonnatural amino acids, is that it does not possess an editing mechanism. The tyrosine analog azatyrosine, L-β-(5-hydroxy-2-pyridyl)-alanine, can convert the Ras-transformed phenotype to normal phenotype, presumably by its incorporation into cellular proteins in place of tyrosine and its inability of being phosphorylated. To understand better this abnormal incorporation, Eco-TyrRS mutants, capable of charging tRNATyr with azatyrosine, were isolated. A library of mutant Eco-tyrS genes was constructed by error prone PCR. Mutant clones (about 1400) were screened for the incorporation of L-[3H]tyrosine or L-[3H]azatyrosine into trichloro-acetic acid precipitable materials. One mutant, carrying the F130S change, showed a 17 fold higher activity for azatyrosine incorporation than the wild-type Eco-TyrRS. According to the Bst-TyrRS structure, Phe130 interacts with Asp182, which receives a H-bond from the hydroxyl of the bound Tyr-AMP (the equivalents of Phe130 and Asp182 in Eco-TyrRS are Ile127 and Asp176 respectively in Bst-TyrRS). The discrimination between tyrosine and azatyrosine, measured in vitro by kcat/KM for the charging of crude E. coli tRNA, decreased from 36 to 19 when going from the wild-type to the F130S mutant.81
In yeast, TyrRS is the aaRS which has the highest discrimination factor between the cognate and noncognate amino acids.82 Sce(cyt)-TyrRS mutants, capable of charging noncanonical amino acids, have been constructed by site-specific mutagenesis of putative active site residues, identified by analogy with Bst-TyrRS. The mutant Sce(cyt)-TyrRS(Y43G), equivalent to Bst-TyrRS(Y34G), was able to utilize 3-substituted tyrosine analogs as substrates for aminoacylation. The catalytic efficiency kcat/KM of Sce(cyt)-TyrRS(Y43G) for aminoacylation with tyrosine was decreased 400 fold as compared to the wild-type. The ability to utilize 3-iodo-L-Tyr was newly generated in this mutant. The mutant TyrRS could serve for site-specific incorporation of new amino acids into proteins.83 In Bst-TyrRS, Tyr34 is a donor of a H-bond to the Oη atom of tyrosine.
The TyrRSs from E. coli, B. subtilis and S. cerevisiae cytoplasm can charge their homologous tRNATyr with D-tyrosine.70,84,85 The resulting D-Tyr-tRNATyr is hydrolyzed by a D-Tyr-tRNATyr-deacylase, which has been identified and characterized in E. coli and S. cerevisiae.85,86 In both organisms, the specificity of the deacylase is not restricted to tRNATyr. In the absence of deacylase, some D-amino acids are toxic to the organism.87 Thus, although TyrRS does not possess an incorporated editing mechanism, the deacylase provides one for some D-amino acids.
Expanding the Genetic Code
An expansion of the genetic code must satisfy three conditions:
- An aaRS N° 21, which specifically charges a tRNA N° 21 to the exclution of the 20 homologous tRNAs, must be introduced into an organism.
- A tRNA N° 21, which is specifically charged by aaRS N° 21 to the exclusion of the 20 homologous aaRSs, and which uses a codon differing from the existing codons, must be introduced into the same organism.
- aaRS N° 21 must specifically recognize, activate and transfer an amino acid N° 21, to the exclusion of the 20 other amino acids.
RajBhandary and coworkers have constructed two couples of aaRS and tRNA which satisfy conditions 1 and 2. One of them is based on Sce(cyt)-TyrRS and an amber suppressor tRNA(CUA), which carries the identity elements Cyt1-Gua72 and Ade73 of the eukaryotic tRNATyr. As described above, plasmids expressing high levels of Sce(cyt)-TyrRS cannot be stably maintained in E. coli, because they mischarge Eco-tRNAPro, which also comprises Cyt1-Gua72 and Ade73. The Sce-tyrS1 gene, coding for Sce(cyt)-TyrRS, was mutagenised by error prone PCR and three mutants were isolated which could be stably expressed in E. coli. The Sce(cyt)-TyrRS mutants quantitatively aminoacylate the tRNA(CUA) in vivo, and show a better discrimination in vitro for the tRNA(CUA) and against Eco-tRNAPro (ref. 69).
Schultz and coworkers have built a couple which satisfies the three conditions above and is based on Mja-TyrRS and Mja-tRNATyr. Mja-TyrRS efficiently aminoacylates an amber suppressor Mja-tRNATyr(CUA), but does not aminoacylate any E. coli tRNA.54,88 The recognition of Mja-tRNATyr(CUA) by the E. coli aaRSs, which is low, was further decreased by the following means. A library of Mja-tRNATyr(CUA) mutants was constructed then panned through a negative selection (absence of aminoacylation by the E. coli aaRSs; no barnase activity) then a positive selection (aminoacylation by Mja-TyrRS, β-lactamase activity) to select a Mja-tRNATyr(CUA, mut) variant.89 To alter the amino acid specificity of Mja-TyrRS, five residues which are located in the vicinity of the Cδ-atom of tyrosine, chosen from the crystal structure of the Bst-TyrRS·Tyr-AMP complex, were first changed into Ala and then randomized to create a library of Mja-TyrRS mutants. The mutants of this library which were able to suppress a nonsense mutation in a nonessential position of the chloramphenicol acetyl transferase gene, in the presence of O-methyl-L-tyrosine but not in its absence, were selected. As a result, a mutant derivative of Mja-TyrRS could incorporate O-methyl-L-tyrosine into proteins by translation of an amber codon, with a fidelity higher than 99 % (ref. 90).
Binding and Charging of tRNATyr Mimics
Sce(cyt)-TyrRS charges the viral RNA of the Brome Mosaic Virus (BMV), or recombinant derivatives of this RNA, with tyrosine. The 3'-OH end of the viral RNA folds into a structure which contains a pseudo-knot and partially mimicks tRNATyr (for a review, see ref. 91). Its aminoacylation depends on nucleotide Ade4 (structural homolog of Ade73 in tRNATyr) and base pair Cyt116-Gua5 (homolog of Cyt1-Gua72). There is no equivalent of the tRNATyr anticodon. Chemical attack experiments, performed on a transcript of 196 nucleotides which can be charged with tyrosine, have indicated that the amino acid acceptor branch of the viral RNA is protected by Sce(cyt)-TyrRS against cleavage by iodine, as well as a hairpin domain which might be located perpendicular to the acceptor branch. This domain, which has no canonical anticodon loop or tyrosine anticodon, could act as an anchor for interaction with TyrRS, leading to a better efficiency of charging.92 Whether or not this hairpin domain makes specific interactions with the synthetase remains unknown.
The mitochondrial TyrRSs from Neurospora crassa, and from Podospora anserina, Ncr(mit)-TyrRS and Pan-(mit)-TyrRS respectively, function in the aminoacylation of the cognate mitochondrial tRNATyr and in the splicing of the group I introns. The splicing activity of the mitochondrial TyrRS seems limited to these two particular organisms. Ncr(mit)-TyrRS is encoded by the cyt-18 gene. It binds to the catalytic core of the group I introns and assists the intron RNA in forming a catalytically active structure. Ncr(mit)-TyrRS splices different group I introns which have little sequence conservation. This sequence comparison has suggested that Ncr(mit)-TyrRS probably recognizes conserved features of secondary and tertiary structure in the intron RNAs. Experiments of chemical attack of the intron RNAs and of Ncr(mit)-tRNATyr, and molecular modeling studies have suggested that Ncr(mit)-TyrRS recognizes a tRNA-like structure of the catalytic core of the group I introns (reviewed in refs. 93, 94).
Comparison of the sequences of Ncr(mit)-TyrRS, Pan(mit)-TyrRS, and other bacterial TyrRSs has shown that the two mitochondrial TyrRSs comprise the four clusters of positively charged residues which are involved in the recognition of tRNATyr (Table 3). Other regions are conserved between Ncr(mit)-TyrRS and Pan(mit)-TyrRS but absent from the other bacterial or mitochondrial TyrRSs. The construction of mutations in Ncr(mit)-TyrRS has helped to further establish its similar modes of interaction with the intron RNAs and tRNATyr, and to characterize the role of its different regions in splicing.95-97 An N-terminal region (residues 41-59), which is absent from the eubacterial TyrRSs and predicted to form an amphipatic a-helix, is required for the splicing activity. It appears to act indirectly, by stabilizing the structure of another TyrRS region which is in direct contact with the intron RNA. The properties of insertion mutations have shown that the aminoacylation activity is not required for the splicing activity. Mutations of the N-terminal extension or of the C-terminal domain have shown that Ncr(mit)-TyrRS favors splicing by different sets of interactions with different group I introns. Thus different functional modes could have evolved from an interaction based on the recognition of a tRNA-like structure.
Eukaryotic TyrRSs and Their Cellular Localization
Several eukaryotic TyrRSs have been studied: from human origin,65 bovine liver,98 rabbit,99 mouse liver,100 wheat germ,101 S. cerevisiae,102 P. carinii,53 etc. The dimeric state of the TyrRSs from bovine liver,98,103 wheat germ,101 and yeast104,105 has been directly established.
Eco-TyrRS, when fused with a mitochondrial import signal, is able to restore respiration of a strain which is defective for this function because of a mutation in Sce(mit)-TyrRS.63 The essential character of Sce(cyt)-TyrRS was shown by gene disruption in a diploid strain.106 The essential character of the Hsa-TyrRS is suggested by the observation that the sera of patients with auto-immune diseases (rheumatoid arthritis and systemic lupus erythematosus) contains antibodies against TyrRS (and other aaRSs), contrary to the sera of healthy patients. The sera of the patients who are ill also contain anti-idiotypic antibodies of IgG type against the auto-antibodies.107
The tRNAs are synthesized in the nucleus and then exported to the cytosol where they are aminoacylated and play their function in translation. In S. cerevisiae, there is a pool of nuclear TyrRS whose import depends on a nuclear localization sequence. The inactivation of this sequence by mutation does not affect the catalytic activity of TyrRS but results in a reduction of its nuclear pool, causes a defect in the export of tRNAs to the cytosol, and results in the nuclear accumulation of tRNATyr, tRNAMet and tRNAAla. The inactivation of TyrRS by a thermosensitive mutation results in the accumulation of tRNATyr, tRNAMet and tRNAIle in the nucleus, at the nonpermissive temperature. Thus, some tRNAs could be exported from the nucleus to the cytosol through an aminoacylation dependent pathway.108,109
Other Properties and Functions of TyrRS
The rabbit liver TyrRS has a TyrRS kinase activity, as the homologous ThrRS has a ThrRS kinase activity.100 The C-terminal domain of the human Hsa-TyrRS is 50% identical to the C-terminal domain of MetRS from C. elegans, at the level of the amino acid sequence, 49% identical to the EMAP II protein, and 43% identical to the Arcp1 protein from S. cerevisiae. These comparisons suggested that the C-terminal domain of Hsa-TyrRS could have a cytokine activity and direct the tRNAs to the active site of the enzyme.65 It was later shown that Hsa-TyrRS can be split into two distinct cytokines. In cell culture under apoptotic conditions, the full length Hsa-TyrRS is secreted and cleaved by an extracellular protease into an N-terminal fragment which is catalytically active for tRNA charging, and a C-terminal fragment. The N-terminal fragment is an interleukin-8 (IL8)-like cytokine, and the C-terminal fragment is an EMAP II-like cytokine. The IL8 activity of the N-terminal fragment depends on a Glu-Leu-Arg motif, which is found in α-chemokines, and is conserved in the TyrRSs from mammals but not from lower eukaryotes. A synthetic heptapeptide, whose sequence is present in the C-terminal domain, has EMAP II-like activity for mononuclear phagocytes and polymorphonuclear leucocytes, but not the homologous peptides from lower eukaryotes. Therefore, the cytokine activities of the split Hsa-TyrRS depend on motifs that are idiosynchratic to the mammalian systems.110,111
The yeast nuclear mutation mgm104-1 leads to slow growth on glucose medium and temperature sensitive loss of mitochondrial DNA. The tyrS1 nuclear gene, coding for Sce(cyt)-TyrRS, can complement the mgm104-1 mutation for these phenotypes when present in two or more copies within the cells. The tyrS1 and mgm104 genes are different since tyrS1 has no mutation in the mgm104-1 mutant allele. These data suggest that tyrS1 has an additional function, which is directly or indirectly involved in the maintenance of the mitochondrial genome.106 Sce(cyt)-TyrRS strongly interacts with the Knr4 protein of S. cerevisiae, as demonstrated by a genetic two-hybrid system and a biochemical pull-down experiment using a GST-TyrRS fusion protein. The Knr4 protein is involved in the regulation of the cell wall assembly in S. cerevisiae. The efficiency of spore formation is drastically reduced in diploid cells, homozygous for a temperature sensitive mutation of the tyrS1 gene or a disruption of the knr4 gene. The physical interaction between the two corresponding proteins might be required for di-tyrosine formation during the sporulation process.112
Bst-TyrRS has been used in hemisynthesis. For example, tyrosine and leucinamide are condensed by TyrRS in the presence of ATP to give tyrosyl-leucinamide, L-Tyr-L-Leu-NH2. TyrRS has no strict specificity for the amino acid derivative used as a substrate and even D-amino acids can be incorporated into peptides in this enzymatic reaction.113 It has thus been possible to synthesize an analgesic neuropeptide, called kyotorphin, H-Tyr-Arg-OH, from tyrosine and arginine. Radioisotope-labeled oligopeptides could be synthesized by this type of reaction and used in receptor binding assays.114
TyrRS and the Classification of Synthetases
TyrRS belongs to class I of the aminoacyl-tRNA synthetases (aaRS) since its catalytic domain has the dinucleotide binding fold and its sequence contains the conserved motifs HIGH and KFGKT.115 In the E. coli system, Fraser and Rich116 have found that the primary site of aminoacylation of tRNATyr by TyrRS is located at the 2'-OH rather than the 3'-OH of the ribose (85% vs 15% of the molecules respectively), as the majority of the class I aaRSs. Sprinzl and Cramer117 have found that both 2'-OH and 3'-OH can be aminoacylated (63% vs 37%). Note that both the 2'-OH and 3'-OH of Ade76 are in proximity of the carbonyl carbon of Tyr-AMP in the model of the Bst-TyrRS·tRNATyr complex.26 In the S. cerevisiae system, tRNATyr can be quantitatively charged both at the 2'-OH and 3'-OH of the Ade76 ribose. The kinetic parameter KM(tRNATyr) of the aminoacylation reaction is the same for tRNATyr-C-C-2_dA, tRNATyr-C-C-3_dA, and tRNATyr-C-C-A. However, Vmax is about 15 times slower for tRNATyr-C-C-2_dA than for the two other tRNATyr species. Thus, tRNATyr is aminoacylated preferentially at the 2'-OH group through a kinetic effect.118 The fact that Vmax but not KM is affected by the presence of 2'-deoxy-Ade76 is consistent with the conclusion that Ade76 is recognized by Bst-TyrRS mainly in the transition state for the transfer reaction.45 The class I aaRSs are mostly monomeric whereas the class II aaRSs are oligomeric. TyrRS and TrpRS are exceptions to this rule since they are obligatory dimers and belong to class I.119
The fact that Bst-TyrRS interacts with tRNATyr according to a class II mode was clearly stated as early as 1993, on the basis of the existing data on the Bst-TyrRS.tRNATyr interaction.59 Because TyrRS belongs unambiguously to class I, its interaction with tRNATyr according to a class II mode was accepted with difficulty. For example, other models or modes of interaction have been proposed.34,44,120-122 The crystal structure of the TyrRS·tRNATyr complex in the T. thermophilus system has completed the demonstration.19 Although tRNATyr can be aminoacylated at either the 2'-OH or the 3'-OH of Ade76, it is preferentially aminoacylated at the 2'-OH for rate reasons, at least in yeast (see above). Thus, TyrRS and PheRS bind their cognate tRNAs according to a class II mode and yet, aminoacylate it preferentially at the 2'-OH, as the canonical aaRSs of class I.
The experimental data showing that TyrRS is an exception among the aaRS, as regards their classification, was developed during the years 1986-1989. 25,26,28,45,58,59,61,123-128 Since then, other exceptions have been observed. For example, there are two types of LysRS, depending on the organism, one belonging to class I and the other to class II.129 In some organisms, tRNACys can be charged by ProRS, which belongs to class II, although its cognate CysRS normally belongs to class I.130-132 It will be interesting to find out how TrpRS interacts with tRNATrp, given its structural homology with TyrRS.133
Acknowledgments
We thank Shamila Na_for critically reading the manuscript, Inaki Guijarro for Figure 1, Stephen Cusack and Eric First for the communication of submitted articles.
References
- 1.
- Burbaum JJ, Schimmel P. Structural relationships and the classification of aminoacyl-tRNA synthetases. J Biol Chem. 1991;266(26):16965–16968. [PubMed: 1894595]
- 2.
- Aravind L, Koonin EV. Novel predicted RNA-binding domains associated with the translation machinery. J Mol Evol. 1999;48(3):291–302. [PubMed: 10093218]
- 3.
- Wolf YI, Aravind L, Grishin NV. et al. Evolution of aminoacyl-tRNA synthetases-analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9(8):689–710. [PubMed: 10447505]
- 4.
- Ribas de Pouplana L, Frugier M, Quinn CL. et al. Evidence that two present-day components needed for the genetic code appeared after nucleated cells separated from eubacteria. Proc Natl Acad Sci USA. 1996;93(1):166–170. [PMC free article: PMC40199] [PubMed: 8552597]
- 5.
- Brown JR, Robb FT, Weiss R. et al. Evidence for the early divergence of tryptophanyl- and tyrosyl-tRNA synthetases. J Mol Evol. 1997;45(1):9–16. [PubMed: 9211729]
- 6.
- Monteilhet C, Blow DM. Binding of tyrosine, adenosine triphosphate and analogues to crystalline tyrosyl transfer RNA synthetase. J Mol Biol. 1978;122(4):407–417. [PubMed: 691047]
- 7.
- Monteilhet C, Blow DM, Brick P. Interaction of crystalline tyrosyl-tRNA synthetase with adenosine, adenosine monophosphate, adenosine triphosphate and pyrophosphate in the presence of tyrosinol. J Mol Biol. 1984;173(4):477–485. [PubMed: 6323720]
- 8.
- Brick P, Blow DM. Crystal structure of a deletion mutant of a tyrosyl-tRNA synthetase complexed with tyrosine. J Mol Biol. 1987;194(2):287–297. [PubMed: 3612807]
- 9.
- Brick P, Bhat TN, Blow DM. Structure of tyrosyl-tRNA synthetase refined at 2.3 Å resolution. Interaction of the enzyme with the tyrosyl adenylate intermediate. J Mol Biol. 1989;208(1):83–98. [PubMed: 2504923]
- 10.
- Waye MM, Winter G, Wilkinson AJ. et al. Deletion mutagenesis using an _M13 splint_: the N-terminal structural domain of tyrosyl-tRNA synthetase (B.stearothermophilus) catalyses the formation of tyrosyl adenylate. EMBO J. 1983;2(10):1827–1829. [PMC free article: PMC555366] [PubMed: 6315404]
- 11.
- Guez-Ivanier V, Bedouelle H. Disordered C-terminal domain of tyrosyl transfer-RNA synthetase: evidence for a folded state. J Mol Biol. 1996;255(1):110–120. [PubMed: 8568859]
- 12.
- Guez V, Nair S, Chaffotte A. et al. The anticodon-binding domain of tyrosyl-tRNA synthetase: state of folding and origin of the crystallographic disorder. Biochemistry. 2000;39(7):1739–1747. [PubMed: 10677223]
- 13.
- Jermutus L, Guez V, Bedouelle H. Disordered C-terminal domain of tyrosyl-tRNA synthetase: secondary structure prediction. Biochimie. 1999;81(3):235–244. [PubMed: 10385005]
- 14.
- Pintar A, Guez V, Castagne C. et al. Secondary structure of the C-terminal domain of the tyrosyl-transfer RNA synthetase from Bacillus stearothermophilus: a novel type of anticodon binding domain? FEBS Lett. 1999;446(1):81–85. [PubMed: 10100619]
- 15.
- Guijarro JI, Pintar A, Prochnicka-Chalufour A. et al. Structure and dynamics of the anticodon-arm binding domain of Bacillus stearothermophilus tyrosyl-tRNA synthetase. Structure. 2002;10(3):311–317. [PubMed: 12005430]
- 16.
- Davies C, Gerstner RB, Draper DE. et al. The crystal structure of ribosomal protein S4 reveals a two-domain molecule with an extensive RNA-binding surface: one domain shows structural homology to the ETS DNA-binding motif. EMBO J. 1998;17(16):4545–4558. [PMC free article: PMC1170785] [PubMed: 9707415]
- 17.
- Markus MA, Gerstner RB, Draper DE. et al. The solution structure of ribosomal protein S4 .41 reveals two subdomains and a positively charged surface that may interact with RNA. EMBO J. 1998;17(16):4559–4571. [PMC free article: PMC1170786] [PubMed: 9707416]
- 18.
- Staker BL, Korber P, Bardwell JC. et al. Structure of Hsp15 reveals a novel RNA-binding motif. EMBO J. 2000;19(4):749–757. [PMC free article: PMC305613] [PubMed: 10675344]
- 19.
- Yaremchuk A, Kriklivyi I, Tukalo M. et al. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J. 2002;21(14):3829–3840. [PMC free article: PMC126118] [PubMed: 12110594]
- 20.
- Park YC, Bedouelle H. Dimeric tyrosyl-tRNA synthetase from Bacillus stearothermophilus unfolds through a monomeric intermediate. A quantitative analysis under equilibrium conditions. J Biol Chem. 1998;273(29):18052–18059. [PubMed: 9660761]
- 21.
- Ward WH, Jones DH, Fersht AR. Protein engineering of homodimeric tyrosyl-tRNA synthetase to produce active heterodimers. J Biol Chem. 1986;261(21):9576–9578. [PubMed: 3733687]
- 22.
- Guez-Ivanier V, Hermann M, Baldwin D. et al. Mapping the stability determinants of bacterial tyrosyl transfer RNA synthetases by an experimental evolutionary approach. J Mol Biol. 1993;234(1):209–221. [PubMed: 8230200]
- 23.
- Park YC, Guez V, Bedouelle H. Experimental evolution of a dense cluster of residues in tyrosyl-tRNA synthetase: quantitative effects on activity, stability and dimerization. J Mol Biol. 1999;286(2):563–577. [PubMed: 9973571]
- 24.
- Carter P, Bedouelle H, Winter G. Construction of heterodimer tyrosyl-tRNA synthetase shows tRNA-Tyr interacts with both subunits. Proc Natl Acad Sci USA. 1986;83(5):1189–1192. [PMC free article: PMC323040] [PubMed: 3006039]
- 25.
- Bedouelle H, Winter G. A model of synthetase/transfer RNA interaction as deduced by protein engineering. Nature. 1986;320(6060):371–373. [PubMed: 3960121]
- 26.
- Labouze E, Bedouelle H. Structural and kinetic bases for the recognition of tRNA-Tyr by tyrosyl-tRNA synthetase. J Mol Biol. 1989;205(4):729–735. [PubMed: 2467006]
- 27.
- Vidal-Cros A, Bedouelle H. Role of residue Glu152 in the discrimination between transfer RNAs by tyrosyl-tRNA synthetase from Bacillus stearothermophilus. J Mol Biol. 1992;223(3):801–810. [PubMed: 1542120]
- 28.
- Bedouelle H, Nageotte R. Macromolecular recognition through electrostatic repulsion. EMBO J. 1995;14(12):2945–2950. [PMC free article: PMC398414] [PubMed: 7796819]
- 29.
- Gaillard C, Bedouelle H. An essential residue in the flexible peptide linking the two idiosynchratic domains of bacterial tyrosyl-tRNA synthetases. Biochemistry. 2001;40(24):7192–7199. [PubMed: 11401566]
- 30.
- Ribas de Pouplana L, Schimmel P. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001;104(2):191–193. [PubMed: 11269237]
- 31.
- Bedouelle H, Guez V, Vidal-Cros A. et al. Overproduction of tyrosyl-tRNA synthetase is toxic to Escherichia coli: a genetic analysis. J Bacteriol. 1990;172(7):3940–3945. [PMC free article: PMC213377] [PubMed: 2113914]
- 32.
- Ward WH, Fersht AR. Asymmetry of tyrosyl-tRNA synthetase in solution. Biochemistry. 1988;27(3):1041–1049. [PubMed: 3365365]
- 33.
- Ward WH, Fersht AR. Tyrosyl-tRNA synthetase acts as an asymmetric dimer in charging tRNA. A rationale for half-of-the-sites activity. Biochemistry. 1988;27(15):5525–5530. [PubMed: 3179266]
- 34.
- Fersht AR. Dissection of the structure and activity of the tyrosyl-tRNA synthetase by site-directed mutagenesis. Biochemistry. 1987;26(25):8031–8037. [PubMed: 3442641]
- 35.
- Fersht AR, Knill-Jones JW, Bedouelle H. et al. Reconstruction by site-directed mutagenesis of the transition state for the activation of tyrosine by the tyrosyl-tRNA synthetase: a mobile loop envelopes the transition state in an induced-fit mechanism. Biochemistry. 1988;27(5):1581–1587. [PubMed: 3284584]
- 36.
- First EA. Catalysis of tRNA aminoacylation by class I and class II aminoacyl-tRNA synthetasesIn: M Sinnott, ed.Comprehensive Biological Catalysis Oxford: Academic Press Ltd.,1998573–607.
- 37.
- Fersht AR, Shi JP, Knill-Jones J. et al. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314(6008):235–238. [PubMed: 3845322]
- 38.
- Lowe G, Tansley G. A stereochemical and positional isotope exchange study of the mechanism of activation of tyrosine by tyrosyl transfer-RNA synthetase from Bacillus stearothermophilus. Tetrahedron. 1984;40(1):113–118.
- 39.
- Leatherbarrow RJ, Fersht AR, Winter G. Transition-state stabilization in the mechanism of tyrosyl-tRNA synthetase revealed by protein engineering. Proc Natl Acad Sci USA. 1985;82(23):7840–7844. [PMC free article: PMC390865] [PubMed: 3865201]
- 40.
- Winter G, Koch GL, Hartley BS. et al. The amino acid sequence of the tyrosyl-tRNA synthetase from Bacillus stearothermophilus. Eur J Biochem. 1983;132(2):383–387. [PubMed: 6840095]
- 41.
- Hountondji C, Dessen P, Blanquet S. Sequence similarities among the family of aminoacyl-tRNA synthetases. Biochimie. 1986;68(9):1071–1078. [PubMed: 3096385]
- 42.
- Hountondji C, Lederer F, Dessen P. et al. Escherichia colityrosyland methionyl-tRNA synthetases display sequence similarity at the binding site for the 3'-end of tRNA. Biochemistry. 1986;25(1):16–21. [PubMed: 3513822]
- 43.
- Avis JM, Day AG, Garcia GA. et al. Reaction of modified and unmodified tRNA(Tyr) substrates with tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Biochemistry. 1993;32(20):5312–5320. [PubMed: 8499435]
- 44.
- Avis JM, Fersht AR. Use of binding energy in catalysis: optimization of rate in a multistep reaction. Biochemistry. 1993;32(20):5321–5326. [PubMed: 8499436]
- 45.
- Xin Y, Li W, Dwyer DS. et al. Correlating amino acid conservation with function in tyrosyl-tRNA synthetase. J Mol Biol. 2000;303(2):287–298. [PubMed: 11023793]
- 46.
- Xin Y, Li W, First EA. The _KMSKS_ motif in tyrosyl-tRNA synthetase participates in the initial binding of tRNA(Tyr). Biochemistry. 2000;39(2):340–347. [PubMed: 10630994]
- 47.
- Xin Y, Li W, First EA. Stabilization of the transition state for the transfer of tyrosine to tRNA(Tyr) by tyrosyl-tRNA synthetase. J Mol Biol. 2000;303(2):299–310. [PubMed: 11023794]
- 48.
- Sherman JM, Rogers K, Rogers MJ. et al. Synthetase competition and tRNA context determine the in vivo identity of tRNA discriminator mutants. J Mol Biol. 1992;228(4):1055–1062. [PubMed: 1474577]
- 49.
- Sherman JM, Rogers MJ, Soll D. Competition of aminoacyl-tRNA synthetases for tRNA ensures the accuracy of aminoacylation. Nucleic Acids Res. 1992;20(11):2847–2852. [PMC free article: PMC336931] [PubMed: 1377381]
- 50.
- McClain WH. Identity of Escherichia colitRNA(Cys) determined by nucleotides in three regions of tRNA tertiary structure. J Biol Chem. 1993;268(26):19398–19402. [PubMed: 8366087]
- 51.
- Hou YM, Schimmel P. Modeling with in vitro kinetic parameters for the elaboration of transfer RNA identity in vivo. Biochemistry. 1989;28(12):4942–4947. [PubMed: 2548595]
- 52.
- Himeno H, Hasegawa T, Ueda T. et al. Conversion of aminoacylation specificity from tRNA(Tyr) to tRNA(Ser) in vitro. Nucleic Acids Res. 1990;18(23):6815–6819. [PMC free article: PMC332736] [PubMed: 2263446]
- 53.
- Quinn CL, Tao N, Schimmel P. Species-specific microhelix aminoacylation by a eukaryotic pathogen tRNA synthetase dependent on a single base pair. Biochemistry. 1995;34(39):12489–12495. [PubMed: 7547995]
- 54.
- Steer BA, Schimmel P. Major anticodon-binding region missing from an archaebacterial tRNA synthetase. J Biol Chem. 1999;274(50):35601–35606. [PubMed: 10585437]
- 55.
- Fechter P, Rudinger-Thirion J, Tukalo M. et al. Major tyrosine identity determinants in Methanococcus jannaschii and Saccharomyces cerevisiae tRNA-Tyr are conserved but expressed differently. Eur J Biochem. 2001;268(3):761–767. [PubMed: 11168416]
- 56.
- Fechter P, Rudinger-Thirion J, Theobald-Dietrich A. et al. Identity of tRNA for yeast tyrosyl-tRNA synthetase: tyrosylation is more sensitive to identity nucleotides than to structural features. Biochemistry. 2000;39(7):1725–1733. [PubMed: 10677221]
- 57.
- Bare LA, Uhlenbeck OC. Specific substitution into the anticodon loop of yeast tyrosine transfer RNA. Biochemistry. 1986;25(19):5825–5830. [PubMed: 3535890]
- 58.
- Nair S, Ribas de Pouplana L, Houman F. et al. Species-specific tRNA recognition in relation to tRNA synthetase contact residues. J Mol Biol. 1997;269(1):1–9. [PubMed: 9192996]
- 59.
- Bedouelle H, Guez-Ivanier V, Nageotte R. Discrimination between transfer-RNAs by tyrosyl-tRNA synthetase. Biochimie. 1993;75(12):1099–1108. [PubMed: 8199245]
- 60.
- Ibba M, Sever S, Praetorius-Ibba M. et al. Transfer RNA identity contributes to transition state stabilization during aminoacyl-tRNA synthesis. Nucleic Acids Res. 1999;27(18):3631–3637. [PMC free article: PMC148616] [PubMed: 10471730]
- 61.
- Lee CP, RajBhandary UL. Mutants of Escherichia coliinitiator tRNA that suppress amber codons in Saccharomyces cerevisiae and are aminoacylated with tyrosine by yeast extracts. Proc Natl Acad Sci USA. 1991;88(24):11378–11382. [PMC free article: PMC53138] [PubMed: 1763051]
- 62.
- Casey JW, Hsu HJ, Getz GS. et al. Transfer RNA genes in mitochondrial DNA of grande (wild-type) yeast. J Mol Biol. 1974;88(4):735–747. [PubMed: 4372363]
- 63.
- Edwards H, Schimmel P. An E. coli aminoacyl-tRNA synthetase can substitute for yeast mitochondrial enzyme function in vivo. Cell. 1987;51(4):643–649. [PubMed: 3315228]
- 64.
- Edwards H, Schimmel P. A bacterial amber suppressor in Saccharomyces cerevisiae is selectively recognized by a bacterial aminoacyl-tRNA synthetase. Mol Cell Biol. 1990;10(4):1633–1641. [PMC free article: PMC362268] [PubMed: 1690848]
- 65.
- Kleeman TA, Wei D, Simpson KL. et al. Human tyrosyl-tRNA synthetase shares amino acid sequence homology with a putative cytokine. J Biol Chem. 1997;272(22):14420–14425. [PubMed: 9162081]
- 66.
- Wakasugi K, Quinn CL, Tao N. et al. Genetic code in evolution: switching species-specific aminoacylation with a peptide transplant. EMBO J. 1998;17(1):297–305. [PMC free article: PMC1170380] [PubMed: 9427763]
- 67.
- Yarus M. Intrinsic precision of aminoacyl-tRNA synthesis enhanced through parallel systems of ligands. Nature - New Biol. 1972;239(91):106–108. [PubMed: 4564911]
- 68.
- Swanson R, Hoben P, Sumner-Smith M. et al. Accuracy of in vivo aminoacylation requires proper balance of tRNA and aminoacyl-tRNA synthetase. Science. 1988;242(4885):1548–1551. [PubMed: 3144042]
- 69.
- Kowal AK, Kohrer C, RajBhandary UL. Twenty-first aminoacyl-tRNA synthetase-suppressor tRNA pairs for possible use in site-specific incorporation of amino acid analogues into proteins in eukaryotes and in eubacteria. Proc Natl Acad Sci USA. 2001;98(5):2268–2273. [PMC free article: PMC30127] [PubMed: 11226228]
- 70.
- Calendar R, Berg P. The catalytic properties of tyrosyl ribonucleic acid synthetases from Escherichia coliand Bacillus subtilis. Biochemistry. 1966;5(5):1690–1695. [PubMed: 4289778]
- 71.
- McArdell JE, Bruton CJ, Atkinson T. The isolation of a peptide from the catalytic domain of Bacillus stearothermophilus tryptophanyl-tRNA synthetase. The interaction of Brown MX-5BR with tyrosyl-tRNA synthetase. Biochem J. 1987;243(3):701–707. [PMC free article: PMC1147915] [PubMed: 3663097]
- 72.
- McArdell JE, Duffield M, Atkinson T. Probing the substrate-binding sites of aminoacyl-tRNA synthetases with the procion dye green HE-4BD. Biochem J. 1989;258(3):715–721. [PMC free article: PMC1138424] [PubMed: 2658972]
- 73.
- Jarvest RL, Berge JM, Houge-Frydrych CS. et al. Interaction of tyrosyl aryl dipeptides with S. aureus tyrosyl tRNA synthetase: inhibition and crystal structure of a complex. Bioorg Med Chem Lett. 1999;9(19):2859–2862. [PubMed: 10522706]
- 74.
- Stefanska AL, Coates NJ, Mensah LM. et al. SB-219383, a novel tyrosyl tRNA synthetase inhibitor from a Micromonospora sp. I. Fermentation, isolation and properties. J Antibiot (Tokyo). 2000;53(4):345–350. [PubMed: 10866215]
- 75.
- Houge-Frydrych CS, Readshaw SA, Bell DJ. SB-219383, a novel tyrosyl tRNA synthetase inhibitor from a Micromonospora sp. II. Structure determination. J Antibiot (Tokyo). 2000;53(4):351–356. [PubMed: 10866216]
- 76.
- Qiu XY, Janson CA, Smith WW. et al. Crystal structure of Staphylococcus aureus tyrosyl-tRNA synthetase in complex with a class of potent and specific inhibitors. Protein Sci. 2001;10(10):2008–2016. [PMC free article: PMC2374228] [PubMed: 11567092]
- 77.
- Jarvest RL, Berge JM, Houge-Frydrych CS. et al. Inhibitors of bacterial tyrosyl-tRNA synthetase: synthesis of carbocyclic analogues of the natural product SB-219383. Bioorg Med Chem Lett. 2001;11(18):2499–2502. [PubMed: 11549455]
- 78.
- Jarvest RL, Berge JM, Brown P. et al. Potent synthetic inhibitors of tyrosyl tRNA synthetase derived from C-pyranosyl analogues of SB-219383. Bioorg Med Chem Lett. 2001;11(5):715–718. [PubMed: 11266176]
- 79.
- Hyde-DeRuyscher R, Paige LA, Christensen DJ. et al. Detection of small-molecule enzyme inhibitors with peptides isolated from phage-displayed combinatorial peptide libraries. Chem Biol. 2000;7(1):17–25. [PubMed: 10662687]
- 80.
- de Prat Gay G, Duckworth HW, Fersht AR. Modification of the amino acid specificity of tyrosyl-tRNA synthetase by protein engineering. FEBS Lett. 1993;318(2):167–171. [PubMed: 8440372]
- 81.
- Hamano-Takaku F, Iwama T, Saito-Yano S. et al. A mutant Escherichia coli tyrosyl-tRNA synthetase utilizes the unnatural amino acid azatyrosine more efficiently than tyrosine. J Biol Chem. 2000;275(51):40324–40328. [PubMed: 11006270]
- 82.
- Freist W, Sternbach H, Pardowitz I. et al. Accuracy of protein biosynthesis: quasi-species nature of proteins and possibility of error catastrophes. J Theor Biol. 1998;193(1):19–38. [PubMed: 9689940]
- 83.
- Ohno S, Yokogawa T, Nishikawa K. Changing the amino acid specificity of yeast tyrosyl-tRNA synthetase by genetic engineering. J Biochem (Tokyo). 2001;130(3):417–423. [PubMed: 11530018]
- 84.
- Calendar R, Berg P. Purification and physical characterization of tyrosyl ribonucleic acid synthetases from Escherichia coliand Bacillus subtilis. Biochemistry. 1966;5(5):1681–1690. [PubMed: 4960133]
- 85.
- Soutourina J, Blanquet S, Plateau P. D-tyrosyl-tRNA(Tyr) metabolism in Saccharomyces cerevisiae. J Biol Chem. 2000;275(16):11626–11630. [PubMed: 10766779]
- 86.
- Soutourina J, Plateau P, Delort F. et al. Functional characterization of the D-Tyr-tRNA-Tyr deacylase from Escherichia coli. J Biol Chem. 1999;274(27):19109–19114. [PubMed: 10383414]
- 87.
- Soutourina J, Plateau P, Blanquet S. Metabolism of D-aminoacyl-tRNAs in Escherichia coliand Saccharomyces cerevisiae cells. J Biol Chem. 2000;275(42):32535–32542. [PubMed: 10918062]
- 88.
- Wang L, Magliery TJ, Liu DR. et al. A new functional suppressor tRNA/aminoacyl-tRNA synthetase pair for the in vivo incorporation of unnatural amino acids into proteins. J Am Chem Soc. 2000;122:5010–5011.
- 89.
- Wang L, Schultz PG. A general approach for the generation of orthogonal tRNAs. Chem Biol. 2001;8(9):883–890. [PubMed: 11564556]
- 90.
- Wang L, Brock A, Herberich B. et al. Expanding the genetic code of Escherichia coli. Science. 2001;292(5516):498–500. [PubMed: 11313494]
- 91.
- Fechter P, Rudinger-Thirion J, Florentz C. et al. Novel features in the tRNA-like world of plant viral RNAs. Cell Mol Life Sci. 2001;58(11):1547–1561. [PubMed: 11706983]
- 92.
- Fechter P, Giege R, Rudinger-Thirion J. Specific tyrosylation of the bulky tRNA-like structure of brome mosaic virus RNA relies solely on identity nucleotides present in its amino acid-accepting domain. J Mol Biol. 2001;309(2):387–399. [PubMed: 11371160]
- 93.
- Lambowitz AM, Caprara MG, Zimmerly S. et al. Group I and Group II ribozymes as RNPs: clues to the past and guides to the future. In: The RNA world. 2nd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory, 1999:451-485.
- 94.
- Caprara MG, Myers CA, Lambowitz AM. Interaction of the Neurospora crassa mitochondrial tyrosyl-tRNA synthetase (CYT-18 protein) with the group I intron P4-P6 domain. Thermodynamic analysis and the role of metal ions. J Mol Biol. 2001;308(2):165–190. [PubMed: 11327760]
- 95.
- Cherniack AD, Garriga G, Kittle Jr JD. et al. Function of Neurospora mitochondrial tyrosyl-tRNA synthetase in RNA splicing requires an idiosyncratic domain not found in other synthetases. Cell. 1990;62(4):745–755. [PubMed: 2143700]
- 96.
- Kittle JrJD, Mohr G, Cranelos JA. et al. The Neurospora mitochondrial tyrosyl-tRNA synthetase is sufficient for group I intron splicing in vitro and uses the carboxy-terminal tRNA-binding domain along with other regions. Genes Dev. 1991;5(6):1009–1021. [PubMed: 1828448]
- 97.
- Mohr G, Rennard R, Cherniack AD. et al. Function of the Neurospora crassa mitochondrial tyrosyl-tRNA synthetase in RNA splicing. Role of the idiosyncratic N-terminal extension and different modes of interaction with different group I introns. J Mol Biol. 2001;307(1):75–92. [PubMed: 11243805]
- 98.
- Korneliuk AI, Kurochkin IV, Matsuka GK. Tyrosyl-tRNA synthetase from the bovine liver. Isolation and physicochemical properties. Mol Biol (Mosk). 1988;22(1):176–186. [PubMed: 3374483]
- 99.
- Wolfson AD, Motorin YA, Ribkinska TI. et al. Purification of mammalian tyrosyl-tRNA synthetase by high-performance liquid chromatography. J Chromatogr. 1990;503(1):277–281. [PubMed: 2341517]
- 100.
- Berg BH. Purification of aminoacyl-tRNA synthetase kinase activities associated with threonyl- and tyrosyl-tRNA synthetases isolated from Bom:NMRI mouse liver. Biochem Mol Biol Int. 1993;29(5):949–958. [PubMed: 8508146]
- 101.
- Quivy JP, Chroboczek J. Tyrosyl-tRNA synthetase from wheat germ. J Biol Chem. 1988;263(30):15277–15281. [PubMed: 3170582]
- 102.
- Chow CM, RajBhandary UL. Saccharomyces cerevisiae cytoplasmic tyrosyl-tRNA synthetase gene. Isolation by complementation of a mutant Escherichia colisuppressor tRNA defective in aminoacylation and sequence analysis. J Biol Chem. 1993;268(17):12855–12863. [PubMed: 8509419]
- 103.
- Gnatenko DV, Korneliuk AI, Kurochkin IV. et al. Isolation and characteristics of functionally active proteolytically modified forms of tyrosyl-tRNA synthetase from bovine liver. Ukr Biokhim Zh. 1991;63(4):61–67. [PubMed: 1949232]
- 104.
- Beikirch H, von der Haar F, Cramer F. Tyrosyl-tRNA synthetase from baker's yeast. Isolation and some properties. Eur J Biochem. 1972;26(2):182–190. [PubMed: 4558241]
- 105.
- Rubelj I, Weygand-Durasevic I, Kucan Z. Evidence for two types of complexes formed by yeast tyrosyl-tRNA synthetase with cognate and noncognate tRNA. Effect of ribonucleoside triphosphates. Eur J Biochem. 1990;193(3):783–788. [PubMed: 2174366]
- 106.
- Guan MX. Cytoplasmic tyrosyl-tRNA synthetase rescues the defect in mitochondrial genome maintenance caused by the nuclear mutation mgm104-1 in the yeast Saccharomyces cerevisiae. Mol Gen Genet. 1997;255(5):525–532. [PubMed: 9294037]
- 107.
- Vartanian OA. Detection of autoantibodies against phenylalanyl-, tyrosyl-, and tryptophanyl-tRNA-synthetase and anti-idiotypic antibodies to it in serum from patients with autoimmune diseases. Mol Biol (Mosk). 1991;25(4):1033–1039. [PubMed: 1795698]
- 108.
- Azad AK, Stanford DR, Sarkar S. et al. Role of nuclear pools of aminoacyl-tRNA synthetases in tRNA nuclear export. Mol Biol Cell. 2001;12(5):1381–1392. [PMC free article: PMC34591] [PubMed: 11359929]
- 109.
- Sarkar S, Azad AK, Hopper AK. Nuclear tRNA aminoacylation and its role in nuclear export of endogenous tRNAs in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1999;96(25):14366–14371. [PMC free article: PMC24442] [PubMed: 10588711]
- 110.
- Wakasugi K, Schimmel P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science. 1999;284(5411):147–151. [PubMed: 10102815]
- 111.
- Wakasugi K, Schimmel P. Highly differentiated motifs responsible for two cytokine activities of a split human tRNA synthetase. J Biol Chem. 1999;274(33):23155–23159. [PubMed: 10438485]
- 112.
- Dagkessamanskaia A, Martin-Yken H, Basmaji F. et al. Interaction of Knr4 protein, a protein involved in cell wall synthesis, with tyrosine tRNA synthetase encoded by TYS1 in Saccharomyces cerevisiae. FEMS Microbiol Lett. 2001;200(1):53–58. [PubMed: 11410349]
- 113.
- Nakajima H, Kitabatake S, Tsurutani R. et al. Dipeptide synthesis catalyzed by aminoacyl-tRNA synthetases from Bacillus stearothermophilus. Int J Pept Protein Res. 1986;28(2):179–185. [PubMed: 3771102]
- 114.
- Kitabatake S, Tsurutani R, Nakajima H. et al. A novel method for the synthesis of kyotorphin, Tyr-Arg, and 3H-Tyr-Arg, catalyzed by tyrosyl-tRNA synthetase from Bacillus stearothermophilus. Pharm Res. 1987;4(2):154–157. [PubMed: 3509140]
- 115.
- Eriani G, Delarue M, Poch O. et al. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347(6289):203–206. [PubMed: 2203971]
- 116.
- Fraser TH, Rich A. Amino acids are not all initially attached to the same position on transfer RNA molecules. Proc Natl Acad Sci USA. 1975;72(8):3044–3048. [PMC free article: PMC432915] [PubMed: 1103136]
- 117.
- Sprinzl M, Cramer F. Site of aminoacylation of tRNAs from Escherichia coliwith respect to the 2'- or 3'-hydroxyl group of the terminal adenosine. Proc Natl Acad Sci USA. 1975;72(8):3049–3053. [PMC free article: PMC432916] [PubMed: 1103137]
- 118.
- Cramer F, Faulhammer H, von der Haar F. et al. Aminoacyl-tRNA synthetases from baker_s yeast: reacting site of enzymatic aminoacylation is not uniform for all tRNAs. FEBS Lett. 1975;56(2):212–214. [PubMed: 1098930]
- 119.
- Cusack S. Eleven down and nine to go. Nat Struct Biol. 1995;2(10):824–831. [PubMed: 7552701]
- 120.
- Jones DH, McMillan AJ, Fersht AR. et al. Reversible dissociation of dimeric tyrosyl-tRNA synthetase by mutagenesis at the subunit interface. Biochemistry. 1985;24(21):5852–5857. [PubMed: 4084496]
- 121.
- Arnez JG, Moras D. Aminoacyl-tRNA synthetase-tRNA recognitionIn: Nagai K, Mattaj IW, eds. RNA-Protein interactions Oxford: Oxford University Press 199453–81.
- 122.
- Fersht AR. Structure and mechanism in protein science. New York: W H Freeman and Company. 1999
- 123.
- Schoemaker HJ, Schimmel PR. Photo-induced joining of a transfer RNA with its cognate aminoacyl-transfer RNA synthetase. J Mol Biol. 1974;84(4):503–513. [PubMed: 4840999]
- 124.
- Perret V, Florentz C, Dreher T. et al. Structural analogies between the 3' tRNA-like structure of brome mosaic virus RNA and yeast tRNA-Tyr revealed by protection studies with yeast tyrosyl-tRNA synthetase. Eur J Biochem. 1989;185(2):331–339. [PubMed: 2684668]
- 125.
- Caprara MG, Lehnert V, Lambowitz AM. et al. A tyrosyl-tRNA synthetase recognizes a conserved tRNA-like structural motif in the group I intron catalytic core. Cell. 1996;87(6):1135–1145. [PubMed: 8978617]
- 126.
- Egorova SP, Iaremchuk AD, Kriklivy IA. et al. Comparative analysis of interaction sites of Thermus thermophilus and Escherichia coli tRNA-Tyr with homologous aminoacyl-tRNA synthetases by means of chemical modification and nuclease hydrolysis. Bioorg Khim. 1998;24(8):593–600. [PubMed: 9784879]
- 127.
- Bedouelle H. Recognition of tRNA(Tyr) by tyrosyl-tRNA synthetase. Biochimie. 1990;72(8):589–598. [PubMed: 2126463]
- 128.
- Perona JJ, Rould MA, Steitz TA. Structural basis for transfer RNA aminoacylation by Escherichia coliglutaminyl-tRNA synthetase. Biochemistry. 1993;32(34):8758–8771. [PubMed: 8364025]
- 129.
- Ibba M, Morgan S, Curnow AW. et al. A euryarchaeal lysyl-tRNA synthetase: resemblance to class I synthetases. Science. 1997;278(5340):1119–1122. [PubMed: 9353192]
- 130.
- Stathopoulos C, Jacquin-Becker C, Becker HD. et al. Methanococcus jannaschii prolyl-cysteinyl-tRNA synthetase possesses overlapping amino acid binding sites. Biochemistry. 2001;40(1):46–52. [PubMed: 11141055]
- 131.
- Stathopoulos C, Li T, Longman R. et al. One polypeptide with two aminoacyl-tRNA synthetase activities. Science. 2000;287(5452):479–482. [PubMed: 10642548]
- 132.
- Lipman RS, Sowers KR, Hou YM. Synthesis of cysteinyl-tRNA(Cys) by a genome that lacks the normal cysteine-tRNA synthetase. Biochemistry. 2000;39(26):7792–7798. [PubMed: 10869184]
- 133.
- Doublie S, Bricogne G, Gilmore C. et al. Tryptophanyl-tRNA synthetase crystal structure reveals an unexpected homology to tyrosyl-tRNA synthetase. Structure. 1995;3(1):17–31. [PubMed: 7743129]
- 134.
- Salazar JC, Zuniga R, Lefimil C. et al. Conserved amino acids near the carboxy terminus of bacterial tyrosyl-tRNA synthetase are involved in tRNA and Tyr-AMP binding. FEBS Lett. 2001;491(3):257–260. [PubMed: 11240138]
- Evolution of TyrRS
- Structure of TyrRS
- Stability of Eubacterial TyrRSs
- Structure of the Complex between TyrRS and tRNATyr
- Asymmetry of TyrRS in Solution
- Kinetics for the Formation of Tyr-AMP
- Kinetics for the Transfer of Tyrosine
- Identity Elements of tRNATyr
- Recognition of tRNATyr and Its Identity Elements by TyrRS
- Species Specificity
- Discrimination between tRNAs
- Inhibitors
- Charging of Noncognate or Nonnatural Amino Acids
- Expanding the Genetic Code
- Binding and Charging of tRNATyr Mimics
- Eukaryotic TyrRSs and Their Cellular Localization
- Other Properties and Functions of TyrRS
- TyrRS and the Classification of Synthetases
- Acknowledgments
- References
- Tyrosyl-tRNA Synthetases - Madame Curie Bioscience DatabaseTyrosyl-tRNA Synthetases - Madame Curie Bioscience Database
- CATHEPSIN PROTEASES IN TOXOPLASMA GONDII - Madame Curie Bioscience DatabaseCATHEPSIN PROTEASES IN TOXOPLASMA GONDII - Madame Curie Bioscience Database
- Molecular Interaction Network of the Hsp90 Chaperone System - Madame Curie Biosc...Molecular Interaction Network of the Hsp90 Chaperone System - Madame Curie Bioscience Database
- Investigating Metastasis Using In Vitro Platforms - Madame Curie Bioscience Data...Investigating Metastasis Using In Vitro Platforms - Madame Curie Bioscience Database
- Molecular Basis of Oncogenesis by NF-κB: From a Bird's Eye View to a RELevant Ro...Molecular Basis of Oncogenesis by NF-κB: From a Bird's Eye View to a RELevant Role in Cancer - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...