Leukodystrophy Overview – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY
Adeline Vanderver, MD, Davide Tonduti, MD, Raphael Schiffmann, MD, Johanna Schmidt, MPH, MGC, CGC, and Marjo S van der Knaap, MD, PhD.
Author Information and AffiliationsInitial Posting: February 6, 2014.
Estimated reading time: 19 minutes
Summary
NOTE: THIS PUBLICATION HAS BEEN RETIRED. THIS ARCHIVAL VERSION IS FOR HISTORICAL REFERENCE ONLY, AND THE INFORMATION MAY BE OUT OF DATE.
Clinical characteristics.
Leukodystrophies are heritable myelin disorders affecting the white matter of the central nervous system with or without peripheral nervous system myelin involvement. Involvement of the white matter tracts almost universally leads to motor involvement that manifests as hypotonia in early childhood and progresses to spasticity over time. This may lead to variable motor impairment, from mild spastic diplegia to severe spastic quadriplegia that limits purposeful movement. In addition, motor dysfunction is likely to significantly impair vital functions including swallowing, chewing, and (in some cases) respiration. Other findings that vary by disorder include extrapyramidal movement disorders (e.g., dystonia and/or dyskinesias), ataxia, seizures, and delay in cognitive development or change in cognitive function over time.
Diagnosis/testing.
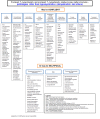
Establishing the specific leukodystrophy present in a given individual usually involves:
Obtaining a medical history and detailed family history
Performing a physical examination and neurologic examination
Review of brain MRI findings:
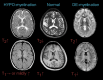
T2-weighted hyperintensity in the white matter is the MRI finding required for diagnosis of a leukodystrophy.
T1-weighted signal may be variable: iso- or hyperintense T1-weighted signal is consistent with a hypomyelinating leukodystrophy; hypointense T1-weighted signal is consistent with a demyelinating leukodystrophy.
Performing specialized laboratory testing, often including molecular genetic testing (either stepwise single-gene testing or use of a multigene panel targeted to the leukodystrophies).
Genetic counseling.
Leukodystrophies with an identified genetic cause may be inherited in an autosomal dominant manner, an autosomal recessive manner, or an X-linked recessive manner. Genetic counseling regarding risk to family members depends on accurate diagnosis, determination of the mode of inheritance in each family, and results of molecular genetic testing. Prenatal testing for pregnancies at increased risk is possible for some types of leukodystrophy if the pathogenic variant(s) in the family are known. Many leukodystrophies are still without an identified genetic cause; once a genetic cause is identified, other inheritance patterns may emerge.
Management.
Treatment of manifestations: Treatment is symptomatic and ideally occurs in a multidisciplinary setting by specialists experienced in the care of persons with a leukodystrophy. Pharmacologic agents are used to manage muscle tone and block neuronal signaling to muscle (chemodenervation). Intensive physical therapy is used to improve mobility and function. Pharmacologic treatment of dystonia and dyskinesias may result in significant functional improvement. Treatment of ataxia, seizures, and cognitive issues is provided in the usual manner, depending on the needs of the individual.
Prevention of primary manifestations: In a few leukodystrophies primary disease manifestations can be prevented by hematopoietic stem cell transplantation (HSCT) or bone marrow transplantation (BMT) early in the disease course.
Surveillance: Routine assessment of growth and nutritional status; physical examination and/or serial x-rays of the hips and spine to monitor for orthopedic complications; and routine history regarding signs and symptoms of seizures.
Agents/circumstances to avoid: Mild head injuries and infection as these may exacerbate disease manifestations.
Evaluation of relatives at risk: When primary prevention of a leukodystrophy is possible (e.g., by HSCT or BMT), it is appropriate to offer testing to asymptomatic at-risk relatives who would benefit from early diagnosis and consideration of early treatment.
Definition of Leukodystrophy
The term "leukodystrophy," as well as associated terms such as "dysmyelination," "demyelination," and "leukoencephalopathy," is applied to a broad group of disorders.
In this GeneReview, the following definition is used: Leukodystrophies are heritable myelin disorders affecting the white matter of the central nervous system with or without peripheral nervous system myelin involvement.
Leukodystrophies share the following findings:
Abnormalities of the glial cell or myelin sheath, such that neuropathology – when known – is characterized primarily by involvement of oligodendrocytes, astrocytes, and other non-neuronal cell types. Of note, in many leukodystrophies the underlying disease mechanism is unknown.
MRI findings (see )
T2-weighted hyperintensity in the white matter must be present.
T1-weighted signal may be variable: iso- or hyperintense T1-weighted signal is consistent with a hypomyelinating leukodystrophy; hypointense T1-weighted signal is consistent with a demyelinating leukodystrophy.
Hypomyelinating leukodystrophy has T2-weighted hyperintensity (↑) and T1-weighted iso- (→) or hyperintensity (↑) of affected white matter. Demyelinating leukodystrophy has T2-weighted hyperintensity (↑) and T1-weighted (more...)
Clinical Manifestations of Leukodystrophies
A number of leukodystrophies that meet the definition used in this GeneReview are listed in Table 1.
Involvement of the white matter tracts almost universally leads to motor involvement that manifests as hypotonia in early childhood and progresses to spasticity over time. This may lead to variable motor impairment, from mild spastic diplegia to severe spastic quadriplegia that limits purposeful movement. In addition, motor dysfunction is likely to significantly impair vital functions including swallowing, chewing, and (in some cases) respiration. Spasticity may result in orthopedic complications such as scoliosis and large joint luxation.
Significant pyramidal dysfunction (i.e., spasticity) may sometimes mask or overshadow the presence of extrapyramidal movement disorders such as dystonia and/or dyskinesias. For example, in MCT8-specific thyroid hormone cell transporter deficiency dystonia is a prominent finding.
Ataxia is a predominant finding in some leukodystrophies and can be disabling; for example, childhood ataxia with central nervous system hypomyelination/vanishing white matter (CACH/VWM) and hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome).
Seizures are an often late manifestation of leukodystrophies, with the exception of rare leukodystrophies (e.g., Alexander disease) in which they are often a presenting feature.
Delay in cognitive development or change in cognitive function over time, while far less pronounced than motor dysfunction, can be common in the child or adult with leukodystrophy. Because progressive loss of cognitive function is slow in the majority of leukodystrophies, dementia is not an early feature.
Prevalence of Leukodystrophies
Epidemiologic data on the frequency of leukodystrophies overall are limited [Heim et al 1997, Bonkowsky et al 2010]. Furthermore, it is difficult to infer from these studies with any certainty the relative frequency of specific leukodystrophies.
Better information on prevalence is available for leukodystrophies that are seen regularly in specialized clinics and in general child neurology practices; these include Alexander disease [van der Knaap et al 1999], X-linked adrenoleukodystrophy [Bezman et al 2001], and metachromatic leukodystrophy.
As disorders such as certain adult-onset-only leukodystrophies and hypomyelinating leukodystrophies become better defined, the heterogeneity of the inherited white matter disorders is increasingly recognized.
It is important to note that in approximately 50% of individuals with a white matter disorder the specific etiology is unknown because of the heterogeneity and complexity of these disorders [Schiffmann & van der Knaap 2009].
Differential Diagnosis of Leukodystrophies
Hereditable disorders with significant white matter involvement that are not leukodystrophies are listed in Table 2 (pdf); they include the following:
Other disorders with significant white matter
involvement include the following:
Acquired CNS myelin disorders that result in demyelination, such as multiple sclerosis and those of infectious, post-infectious, and autoimmune etiology. Although not heritable in a Mendelian fashion, some of these disorders may have multifactorial etiologies that include a genetic predisposition.
Disorders such as acute disseminated encephalomyelitis, multiple sclerosis, and neuromyelitis optica typically differ from heritable white matter disorders by their abrupt onset and multiphasic presentations. Brain MRI abnormalities are also more likely to be multifocal and patchy [
Schiffmann & van der Knaap 2009], with variation or even improvement over time and with treatment.
Toxic injuries of the myelin, such as those seen in heroin abuse or methotrexate-related toxicity
Central nervous system injury, particularly in the perinatal period, which can result in significant white matter signal abnormalities that are typically irregular, and may result in loss of white matter volume
Non-genetic vascular insults
Evaluation Strategy for an Individual with a Leukodystrophy
Once a leukodystrophy is considered in an individual, the following approach can be used to determine the specific leukodystrophy to aid in discussions of prognosis and genetic counseling.
Establishing the specific leukodystrophy (Table 1) present in a given individual usually involves obtaining a medical history and detailed family history; performing a physical examination and neurologic examination; review of brain MRI findings; and specialized laboratory testing, often including molecular genetic testing.
Note: Adherence to the diagnostic approach discussed in this section notwithstanding, a specific diagnosis cannot be established in a clinical (i.e., not research) setting in a significant number of individuals with a leukodystrophy [van der Knaap et al 1999, Schiffmann & van der Knaap 2009].
Medical History
A history of certain clinical features may be helpful in identifying a specific leukodystrophy; however, in the majority of cases, only nonspecific loss of function (primarily motor) occurs and medical history alone does not provide insight into a specific diagnosis.
In the hypomyelinating leukodystrophies helpful diagnostic clues may, for example, be the following:
In the demyelinating leukodystrophies helpful diagnostic clues may, for example, be the following:
Family History
A detailed three-generation family history should be compiled, focusing on individuals with hypotonia, spasticity, dystonia, seizures, ataxia, and/or delay in cognitive development or change in cognitive function over time.
Because most leukodystrophies are autosomal recessive, special attention to parental consanguinity and medical problems in sibs is warranted.
Evaluation of relatives and/or review of their medical records may be needed.
Physical Examination and Neurologic Examination
In most instances physical findings do not suggest a specific diagnosis; however, certain findings may direct the reader to further explore specific underlying etiologies:
Brain MRI Findings
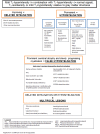
Step 2
Within the hypomyelinating and demyelinating leukodystrophies, determine if patterns of involvement suggest specific diagnoses.
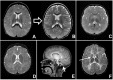
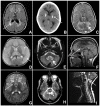
Hypomyelinating leukodystrophy. Note specific patterns on neuroimaging that include the following ():
Hypomyelinating leukodystrophy – patterns on MRI A→B. Improvement of myelination over time in MCT8-specific thyroid hormone cell transporter deficiency seen in a male with documented SLC16A2 (MCT8) pathogenic variants at age two years (more...)
Severe atrophy of cortical gray matter (variably seen in primary neuronal disorders as well as a limited number of classic leukodystrophies; see )
Basal ganglia involvement (e.g., in HABC syndrome; see )
Demyelinating leukodystrophy
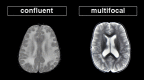
Confluent white matter lesions are extensive white matter abnormalities in significant portions of the brain, often affecting specific regions or tracts, although not necessarily perfectly symmetrically. Multifocal white matter lesions are more discrete, (more...)
Specific patterns on neuroimaging in confluent disorders include ():
Demyelinating leukodystrophy – patterns on MRI A. Diffuse cerebral involvement in an individual with megalencephalic leukodystrophy with subcortical cysts
Cerebellar/cerebellar peduncle involvement (e.g., in
AD adult-onset leukodystrophy (ADLD) with involvement of the middle cerebellar peduncles; see )
Diagnostic algorithm. For algorithms based on MRI findings, see (demyelinating and other conditions) and (hypomyelinating conditions) [Schiffmann & van der Knaap 2009].
Algorithm Part 1: Demyelinating and other conditions Adapted from Schiffmann & van der Knaap [2009]
Algorithm Part 2: Hypomyelinating conditions Adapted from Schiffmann & van der Knaap [2009]
Step 3
Look for the following associated features which, in addition to the pattern of findings on brain MRI, can assist in recognition of a specific leukodystrophy:
Leukoencephalopathy with macrocephaly(
Table 6)
Cerebellar abnormalities (
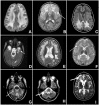
Table 8) seen in the dentate nucleus (e.g., in L-2-hydroxyglutaric aciduria; see )
Non-calcifying basal ganglia lesions (
Table 10) (e.g., in
Alexander disease; see , signal abnormality of the basal ganglia and frontal white matter)
Spinal cord involvement (
Table 12) (seen in many disorders, including
LBSL; see , an example of involvement of tracts within the spinal cord)
Features associated with specific leukodystrophies A. White matter rarefaction and cysts on FLAIR imaging in vanishing white matter disease; arrow indicates cystic rarefaction within abnormal white matter.
Specialized Laboratory Testing (Including Molecular Genetic Testing)
If the findings on brain MRI are consistent with a specific leukodystrophy, consider biochemical or molecular genetic testing for that disorder (Table 1). Note: Molecular genetic testing may be performed either as single-gene testing in a stepwise fashion based on the information gained during the evaluation or, if available, as a multigene panel.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Table 1.
Leukodystrophies Meeting Strict Diagnostic Criteria
View in own window
| Name of Disorder | Mode of Inheritance | Gene 1 | Biochemical Testing / Other |
|---|
| 18q deletion syndrome | Most often de novo deletion; may be inherited | | Chromosome analysis for 18q microdeletion involving MBP |
| Adult polyglucosan body disease (APBD) | AR |
GBE1
| Histopathologic examination of muscle, nerve, axillary skin: pathologic polyglucosan accumulation |
| Aicardi-Goutières syndrome (AGS) | Usually AR; may be AD |
TREX1
RNASEH2A
RNASEH2B
RNASEH2C
SAMHD1
ADAR
| CSF analysis: lymphocytosis, ↑ interferon-α, ↑ pterins |
|
Alexander disease
| AD |
GFAP
| |
| AD adult-onset leukodystrophy (ADLD) | AD |
LMNB1
| |
| Cerebroretinal microangiopathy w/calcifications & cysts (CRMCC) 2 | Likely AR | | Clinical & neuroradiologic features |
|
Canavan disease
| AR |
ASPA
| In urine, plasma, CSF, & amniotic fluid: ↑ N-acetylaspartic acid in urine;
In skin fibroblasts: deficient aspartoacylase enzyme activity |
| Cerebrotendinous xanthomatosis (CTX) | AR |
CYP27A1
| In plasma & CSF: ↑ cholestanol concentration, ↓ chenodeoxycholic acid;
In bile, urine, plasma: ↑ concentration bile alcohols & glyconjugates;
In fibroblasts, liver, leukocytes: ↓ sterol 27-hydroxylase activity |
| Childhood ataxia w/CNS hypomyelination / vanishing white matter (CACH/VWM) | AR |
EIF2B1-5
| |
| Free sialic acid storage disorders 3 | AR |
SLC17A5
| In urine, fibroblast, lysosomes: ↑ free sialic acid |
| Fucosidosis | AR |
FUCA1
| On urinary oligosaccharide assay: ↑ fucose-containing glycoconjugates;
In leukocytes or fibroblasts: deficient α-fucosidase activity |
| Hypomyelination w/atrophy of the basal ganglia & cerebellum (H-ABC) | Likely AD |
TUBB4A
| Clinical & neuroradiologic features |
| Hypomyelination and congenital cataract (HCC) | AR |
FAM126A
| |
|
Krabbe disease
| AR | GALC
See footnote 4 | In leukocytes or fibroblasts: deficient galactocerebrosidase activity |
| L-2-hydroxyglutaric aciduria | AR |
L2HGDH
| In plasma, urine, CSF: ↑ concentration of L-2-hydroxyglutaric acid (and lysine) |
| Leukoencephalopathy w/brain stem & spinal cord involvement & lactate elevation (LBSL) | AR |
DARS2
| |
| Leukoencephalopathy w/thalamus and brain stem involvement & lactate elevation (LTBL) | AR |
EARS2
| |
| Megalencephalic leukodystrophy w/subcortical cysts (MLC) | AR |
MLC1
HEPACAM (MLC2)
| |
| Metachromatic leukodystrophy (MLD) | AR |
ARSA
| In leukocytes, fibroblasts: ↓ arylsulfatase A activity;
In urine: ↑ sulfatides |
| PSAP-related MLD 5 | |
PSAP
| In leukocytes, fibroblasts: normal arylsulfatase A activity;
In urine: ↑ sulfatides |
| Multiple sulfatase deficiency (MSD) | |
SUMF1
| ↓ activity of other sulfatases;
In urine: ↑ mucopolisaccharides, ↑ urinary sulfatides |
| Hereditary diffuse leukoencephalopathy w/spheroids (HDLS) 6 | AD |
CSF1R
| |
| Oculodentodigital dysplasia (ODDD) | Usually AD; may be AR |
GJA1
| |
| Pelizaeus-Merzbacher disease (PMD) | XL |
PLP1
| |
| Pelizaeus-Merzbacher-like disease 1 (PMLD1) | AR |
GJC2
| |
| Zellweger spectrum disorder (PBD, ZSD) 7 | AR | PEX genes | Plasma VLCFA, phytanic & pristanic acid, plasma & urine concentration of pipecolic acid & bile acids aid to distinguish different forms of peroxisomal disorders |
| Pol III-related leukodystrophies 8 | AR |
POLR3A
POLR3B
| |
| RNAse T2-deficient leukoencephalopathy | AR |
RNASET2
| |
| Single-enzyme deficiencies of peroxisomal fatty acid beta oxidation 9 | AR | Dibifunctional protein deficiency: HSD17B4 | Plasma VLCFA, phytanic & pristanic acid, plasma & urine concentration of pipecolic acid & bile acids aid to distinguish different forms of peroxisomal disorders |
| Peroxisomal acyl-CoA-oxidase deficiency: ACOX1 |
| SCPx deficiency: SCP2 |
| Sjögren-Larsson syndrome | AR |
ALDH3A2
| In urine: abnormal metabolites of leukotriene B4;
In cultured skin fibroblasts, leukocytes: deficiency of fatty aldehyde dehydrogenase activity (FALDH) and/or of fatty alcohol:NAD oxidoreductase (FAO) |
| SOX10-associated disorders | AD |
SOX10
| |
| X-linked adrenoleukodystrophy (X-ALD) | XL |
ABCD1
| On plasma VLCFA assay: C26:0, ↑ ratio of C24:0 to C22:0, ↑ ratio of C26:0 to C22:0 |
AD = autosomal dominant; AR = autosomal recessive; VLCFA = very long-chain fatty acid; XL = X-linked
Disorders listed in alphabetic order; naming as per GeneReviews
- 1.
Genetic testing is available for many of these genes.
- 2.
This disorder now appears to be distinct from Coats plus caused by pathogenic variants in CTC1, encoding conserved telomere maintenance component 1.
- 3.
Includes Salla disease; infantile sialic acid storage disease, intermediate form
- 4.
Defects in PSAP causing a deficiency in the activator protein of SapA-d essential for the action of GALC have been reported.
- 5.
Pathogenic variants in PSAP result in deficiency in SapB-d, an activator protein essential for ARSA activity.
- 6.
Also known as adult-onset leukodystrophy w/ neuroaxonal spheroids & pigmented glia; may include hereditary diffuse; pigmentary type of orthochromatic leukodystrophy w/pigmented glia (POLD)
- 7.
Includes neonatal adrenoleukodystrophy; infantile Refsum disease
- 8.
Includes hypomyelination, hypodontia, hypogonadotropic hypogonadism (4H syndrome); ataxia, delayed dentition, and hypomyelination (ADDH); tremor-ataxia with central hypomyelination (TACH); leukodystrophy with oligodontia (LO); and hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum (HCAHC).
- 9.
Includes D-bifunctional protein (DBP) deficiency; sterol carrier protein-2 (SCPx) deficiency; peroxisomal acyl-CoA-oxidase deficiency
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Leukodystrophies with an identified genetic cause may be inherited in an autosomal dominant manner, an autosomal recessive manner, or an X-linked recessive manner; other inheritance patterns may be identified as more genetic causes of leukodystrophy are discovered.
If a proband has a specific syndrome associated with a leukodystrophy, counseling for that condition is indicated.
Risk to Family Members
Autosomal Dominant Inheritance
Parents of a proband
Most individuals diagnosed as having autosomal dominant leukodystrophy have an affected parent, although occasionally the family history is negative.
Family history may appear to be negative because of early death of a parent, failure to recognize autosomal dominant leukodystrophy in family members, late onset in a parent, reduced penetrance of the mutated allele in an asymptomatic parent, or a de novo pathogenic variant.
Sibs of a proband
The risk to sibs depends on the genetic status of the proband's parents.
If one of the proband's parents has a mutated allele, the risk to the sibs of inheriting the mutated allele is 50%.
Offspring of a proband. Individuals with autosomal dominant leukodystrophy have a 50% chance of transmitting the mutated allele to each child.
Autosomal Recessive Inheritance
Parents of a proband
Sibs of a proband
At conception, each sib of a proband has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Once an at-risk sib is known to be unaffected, the chance of his/her being a carrier is 2/3.
Offspring of a proband. All offspring are obligate carriers.
X-Linked Inheritance
Parents of a proband
The father of an affected male will not have the disease nor will he be a carrier of the pathogenic variant.
Women who have an affected son and another affected male relative are obligate heterozygotes. These females may be affected, sometimes with only certain features of the disease, or with milder symptoms. Note: If a woman has more than one affected child and no other affected relatives and if the pathogenic variant cannot be detected in her leukocyte DNA, she has germline mosaicism. Rarely, the unaffected father of an affected female may have germline mosaicism.
A mother of an affected female who has a pathogenic variant may have favorably skewed X-chromosome inactivation that results in her being unaffected or mildly affected.
An individual who is the only affected family member (i.e., a simplex case) may have a de novo pathogenic variant.
Sibs of a proband. The risk to sib depends on the carrier status of the mother:
If the mother of the proband has a pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Male sibs who inherit the variant will be affected; female sibs who inherit the variant will be carriers and will usually not be affected.
If the proband represents a simplex case (i.e., a single occurrence in a family) and if the pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is low but greater than that of the general population because of the possibility of maternal germline mosaicism.
Offspring of a proband
Daughters of an affected male will be obligate carriers and may or may not be affected; none of his sons will be affected.
Each child of an affected female has a 50% chance of inheriting the pathogenic variant.
Because of possible skewing of X-chromosome inactivation, variable phenotypes in females are possible.
Other family members of a proband. The risk to other family members depends on the genetic status of the proband's parents.
Prenatal Testing and Preimplantation Genetic Testing
Once the pathogenic variant(s) have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for leukodystrophy are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Medical Home Portal
The Parents & Families section of the Medical Home Portal provides information and resources to help families learn how to better care for a child with chronic and complex conditions and to become more effective partners in their child’s care.
Department of Pediatrics University of Utah
P.O. Box 581289
Salt Lake City UT 84158
Phone: 801-213-3920
Email: info@medicalhomeportal.org
Myelin Disorders Bioregistry Project
Email: myelindisorders@cnmc.org
Management
Treatment of Manifestations
Although the underlying mechanisms of leukodystrophies are diverse, many manifestations are similar across this group of disorders. In the great majority of cases, primary treatment is not possible, but management of symptoms can improve the comfort and care of individuals with these complex disorders.
Ideally, the child or adult with a leukodystrophy is managed in a multidisciplinary setting by providers experienced in the care of persons with a leukodystrophy.
Spasticity. Pharmacologic agents are used to manage muscle tone and block neuronal signaling to muscle (chemodenervaton). Intensive physical therapy is used to improve mobility and function.
Extrapyramidal manifestations. Dystonia and dyskinesias may cause significant disability; pharmacologic treatment may result in significant functional improvement.
Ataxia. No specific treatment of ataxia exists, although rehabilitative measures can be of great assistance.
Seizures. Seizures should be treated with typical anticonvulsants and are rarely refractory, except on occasion at the end of life.
Cognitive developmental delay / encephalopathy. It is important to advocate for persons with a leukodystrophy in school or at work to avoid limitations related to their motor disabilities. Augmentative communication may be used to address speech deficits. Accommodations for cognitive delays and fine motor disabilities should be used as needed.
Orthopedic. Attention should be given to the prevention and treatment of orthopedic problems, such as hip dislocation and scoliosis.
Feeding. Swallowing dysfunction and pulmonary problems resulting from the increased risk of aspiration are common as the disease progresses. Decreased nutritional intake and failure to thrive may also occur. The decision to place a gastrostomy tube for nutrition is based on the overall health status of the individual, expected disease course, and family and patient wishes.
Prevention of Primary Manifestations
Primary disease manifestations can be prevented in a few of the leukodystrophies: in X-linked adrenoleukodystrophy, Krabbe disease, and metachromatic leukodystrophy, for example, hematopoietic stem cell transplantation (HSCT) or bone marrow transplantation (BMT) may be beneficial if performed early in the disease course. Patients with these disorders should be referred to specialized centers for consideration of HSCT or BMT [Eichler et al 2009].
Surveillance
Standard surveillance includes the following:
Routine measurement of weight and height to assess growth and nutritional status
Physical examination and/or serial x-rays of the hips and spine to monitor for orthopedic complications
Routine history regarding signs and symptoms of seizures
Certain disorders require specialized surveillance; for example, monitoring for the development of hydrocephalus in Alexander disease.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Chapter Notes
Revision History
30 January 2020 (ma) Retired chapter: Phenotype is too broad.
6 February 2014 (me) Review posted live
14 February 2012 (av) Original submission